Dr_bieger_neurostress-guide-juli.2009-kurz201
PD Dr med WP Bieger
NeuroStress Guide
EINLEITUNG
Der vorliegende NeuroScience-Guide ist als Anleitung für Patienten, Ärzte und Therapeuten
gedacht. Er soll einen Einblick in die Funktionsweise des Neuroendokriniums und in die
Grundlagen neuroendokriner Funktionsstörungen und deren Behandlung vermitteln. Die von uns
entwickelte „NeuroStress"-Diagnostik wird vorgestellt und physiologische Behandlungsformen
besprochen. Schon lange gibt es hochwirksame Substanzen für die natürliche Behandlung
psychoneurovegetativer Störungen, die allerdings durch die Entwicklung der modernen
Psychopharmaka verdrängt wurden. Die unbefriedigenden Ergebnisse der Antidepressiva haben
die traditionellen Behandlungskonzepte jedoch wieder ins Bewusstsein gerückt. Unser aktuelles
ganzheitliches diagnostisch-therapeutisches Konzept greift die bewährten Verfahren auf und
verbindet sie mit innovativen Diagnose- und Behandlungsformen aus den USA.
Eingangsüberlegungen:
1. Die Zahl neurovegetativer Störungen und psychischer Krankheiten nimmt weltweit stark zu.
Damit auch die Nachfrage nach neuen diagnostischen Möglichkeiten und effzienten, gut
verträglichen Behandlungen. Seit Jahren steigt die Zahl psychischer Störungen in den westlichen
Industrieländern. Man geht davon aus, dass bis zu 60% der Krankheitsfälle in der täglichen
ärztlichen Praxis psychischer Natur sind bzw. eng mit psychischen Belastungen verbunden sind.
Schon heute entfallen viele Krankheitstage auf psychische Störungen, ihre Zahl nimmt ständig
zu, während die Gesamtzahl krankheitsbedingter Fehltage seit Jahren zurückgeht. Besonders
gravierend ist die Zunahme der Depressionen. Während Herz-Kreislauferkrankungen,
Herzinfarkt, sogar die häufigsten Krebserkrankungen (Lungen-, Brust- und Prostatakrebs) seit
einigen Jahren abnehmende Tendenz zeigen, nimmt der Anteil von Depressionen ständig zu. Die
WHO geht in einem ihrer jüngsten Gesundheitsberichte (2006) davon aus, dass bereits in den
nächsten 5-10 Jahren Depressionen die zweithäufigste medizinische Krankheitsursache
überhaupt sein werden.
2. Psychopharmaka werden heute in enormem Maße eingesetzt, sie sind mit >65 Mrd € die
umsatzstärkste pharmazeutische Präparategruppe. Ihre Wirksamkeit, vor allem die der
Antidepressiva, ist jedoch begrenzt. Die Nebenwirkungen sind zahlreich und zum Teil
lebensbedrohlich. Immer wieder werden Zweifel am Aussagewert von Psychopharmakastudien
geäußert, die Publikation von klinischen Studien mit Antidepressiva erfolgt offensichtlich nach
willkürlichen Kriterien (NEJM, 2008). In einer kürzlichen Metaanalyse wurde die fehende
Wirksamkeit von Antidepressiva bei leichten bis mittelschweren Depressionen konstatiert (PloS
Medicine, 2008). Nur bei schweren Depressionen findet sich ein Vorteil von Antidepressiva
gegenüber Placebos.
3. Die Neurobiologie psychischer Störungen wird seit Jahren intensiv untersucht, neue
Untersuchungsverfahren wie MRT, PET, SPECT, Immunologie oder Molekulargenetik haben das
Verständnis der zentralen Prozesse enorm verbessert. Neue Diagnose- und
Behandlungsverfahren können daher stärker auf Wissen und weniger auf Zufallsbeobachtungen
wie bisher basiert werden.
STRESS
Als eines der zentralen Geschehen gilt chronischer Stress, der vielfach zu bleibenden
psychovegetativen Störungen führt. Ein kürzlich erschienenes Buch des Bonner Psychiaters Prof.
Benkert (2006) hat den Begriff der „
StressDepression" geprägt und damit „Stress" als ganz
wesentlichen Grund für psychische Störungen einschließlich Depressionen in den Mittelpunkt
gestellt. Die medizinischen Konsequenzen dieser Neudefinition von Erkrankungen sind erheblich. War z.B. der Herzinfarkt lange Zeit fast ausschließlich eine Domäne der Arterioskleroseforschung und im engeren Sinne ein
Cholesterin-Problem, so verändert sich inzwischen die Sicht der Herz-Kreislauf-Erkrankungen deutlich. Natürlich spielen die bekannten Risikofaktoren wie falsche Ernährung, Adipositas, Bluthochruck, Rauchen, Cholesterin, LDL/HDL nach wie vor eine bedeutende Rolle, aber nur maximal 50% der Herzinfarkte hängen mit hohem Cholesterin zusammen, häufig sind gar keine Risikofaktoren feststellbar und der Herzinfarkt kommt unvorbereitet, sozusagen „aus heiterem Himmel". Heute wissen wir, dass Stress, bzw. die gravierenden gesundheitlichen Veränderungen durch anhaltenden Stress an dem Krankheitsprozess beteiligt sind und sogar unmittelbare Auslöser des Herzinfarkts sein können und dass die Vermeidung oder zumindest Verringerung von Stress eine der wichtigsten Maßnahmen zur Vorbeugung von Komplikationen bei erhöhtem Infarktrisiko ist. Bei der Fußballweltmeisterschaft 2006 in Deutschland stieg z.B. die Zahl der Herzinfarkte, die in Münchner Klinken eingeliefert wurden, bis zum Dreifachen des mehrjährigen Durchschnitts immer dann an, wenn die deutsche Mannschaft spielte (Wilbert-Lampen, 2008). Bei wichtigen Spielen anderer Mannschaften war die Infarktrate dagegen gegenüber den Vorjahren nicht nennenswert erhöht. Psychosoziale Faktoren wiegen insgesamt schwerer für das Herzinfarktrisiko als alle bekannten wichtigen Faktoren (Adipositas, Rauchen, Diabetes, Hochdruck) zusammen (Interheart Study; Yusuf, 2004). Nach einem Herzinfarkt ist die Stressverminderung die zweiteffektivste Maßnahme zur Vermeidung erneuter kardialer Komplikationen, nach der Ernährungsumstellung und noch vor fettarmer Kost, mediterraner Ernährungsweise, verstärkter körperlicher Aktivität und Beendigung des Rauchens (Kolenda, 2003). Nicht nur Herz-Kreislauf-Erkrankungen, auch eine wachsende Zahl weiterer gesundheitlicher Komplikationen sind Folgen von anhaltendem, chronischem Stress und den daraus entstehenden Störungen der neurovegetativen, hormonellen und metabolischen Regulation, wobei das Zentralnervensystem, das sympathische und parasympathische Nervensystem, die zentrale Stresshormonachse, zahlreiche weitere Hormonsysteme wie u.a. die Nebenniere und die Schilddrüse und darüber hinaus das Immunsystem involviert sind (Lupien, 2009; Shonkoff, 2009; McEwen, 2008). Stress erhöht das Risiko für Infektionen (Grippe, Erkältungen), die Reaktivierung latenter Virusinfektionen (Gürtelrose), die Entwicklung von Autoimmunerkrankungen und sowohl für die Entstehung als auch die Ausbreitung von Krebserkrankungen, was unmittelbar mit den immunologischen Auswirkungen von anhaltendem Stress zusammenhängt. Im Begriff der
PsychoNeuroEndokrinoImmunologie werden inzwischen die Wechselbeziehungen zwischen Nervensystem, Endokrinium, Immunsystem und Psyche zusammengefasst. Die heutige Vorstellung von „Stress" wurde bereits um 1930 von Selye geprägt, der die klassische Vorstellung erweiterte und als „Stresssyndrom" die Adaptation an jede Art von physischen oder emotionalen Kräften, die die Homöostase des Organismus fordern, definierte. Stress wird heute als Zustand echter oder als solcher wahrgenommener Störung der körperlichen Homöostase verstanden. Stress ist nicht nur Ausdruck von privaten oder sozialen, emotionalen oder psychischen Belastungen (
psychischer und mentaler Stress)
. Auslöser von Stress, also „
Stressoren", sind genauso Infektionen, Verletzungen, Entzündungen, auch und vor allem die zahlreichen normalen, physiologischen Abläufe wie die Nahrungsaufnahme, deren Energiegehalt und Energiedichte den Stoffwechsel belasten und im Übermaß einen erheblichen
metabolischen Stress bedingen, da jede aufgenommene Kalorie die metabolische Aktivität erhöht und über die Steigerung der mitochondrialen Energiebereitstellung auch vermehr oxidative Metaboliten anfallen (ROS,
oxidativer Stress); auch die Zusammensetzung der Nahrung, der Anteil an gesättigten/ ungesättigten Fettsäuren, das Verhältnis von Fetten, Kohlehydraten und Eiweiß in der Nahrung haben Einfluss; ebenso Bewegungsmangel, der die Stressadaptation beeinträchtigt, umgekehrt auch übermäßige körperliche Aktivität (
physischer Stress). Umweltnoxen, Schadstoffe, Schwermetalle, Chemikalien in Nahrungsmitteln (Konservierungsstoffe, etc), Pflegemittel ebenso wie Medikamente bewirken auf zellulärer Ebene vermehrte Belastung (
chemischer Stress); Lärmbelastung, Reizüberflutung, übermäßiger Fernseh-/EDV-Konsum, Schlafmangel (
sensorischer Stress), schulische, berufliche Belastungen, Freizeit-Belastungen, die hohe Arbeitsintensität, beruflicher Konkurrenzdruck, mangelnde Anerkennung, Mobbing,
soziale Vereinzelung, Einsamkeit, familiäre Schicksalsschläge, Partnerkonflikte, sexueller Missbrauch, Ängste (
psychischer und sozialer Stress). All diese Stressoren aktivieren über einen einheitlichen, zentralen Mechanismus das körpereigene Stressbewältigungsprogramm, bestehend aus hormonellen Faktoren, Anteilen des zentralen und autonomen Nervensystems und dem immuninflammatorischen Komplex. Die Stresstoleranz, die Fähigkeit auch stärkere und lang anhaltende Belastungen ausgleichen, ist individuell unterschiedlich ausgeprägt. Bei einer wachsenden Zahl von Menschen führt lang anhaltender Stress zu gesundheitlichen Störungen, die nicht selten in Krankheiten übergehen. Dabei sind es seltener einzelne, extreme Stresssituationen wie das
Postraumatische Stressyndrom (PTSD/PTSS) oder das „
Gofkriegssyndrom", sondern eher die kumulative Langzeitwirkung von Stressoren. Das Auftreten und Ausmaß gesundheitlicher Komplikationen ist nicht nur von der Stressdauer und -intensität abhängig sondern eben auch von individuell disponierenden Faktoren (familiäre Besonderheiten, angeborene Risikofaktoren), die maßgeblich für die individuelle Toleranzschwelle sind (Su, 2009). Zu den häufigsten gesundheitlichen Auswirkungen zählen
Schlaflosigkeit, Leistungsabfall, Nervosität, Konzentrationsschwäche, Motivationsverlust bis zum Burn-out, Gedächtnisstörungen, auch
Essstörungen, Überempfindlichkeitsreaktionen, ggf. auch Schmerzen, Stimmungsschwankungen, Ängste und Depressionen. Die wichtigsten Erkrankungen, die auf anhaltenden Veränderungen des neuroendokrin-immunologischen Steady-state basieren, sind im Folgenden aufgelistet (Häufigkeit in %):
CFS/Chronisches Müdigkeitssyndrom
Fatigue (Müdigkeit/Erschöpfbarkeit)
FMS/Fibromyalgie
Übergewicht/Adipositas
Kohlehydrat-Heißhunger
Appetitstörungen
Bipolare Depression
Burn-out Syndrom
Schlafstörungen
MCS/Multiple Chemische Sensitivität
PMS/Prämenstruelles Syndrom
Menopausebeschwerden
Irritables Colon (Reizdarm)
RLS/Restless Leg-Syndrom
PHYSIOLOGIE DER STRESSBEWÄLTIGUNG
Der Organismus verfügt über ein äußerst komplexes, flexibles und leistungsfähiges Stress-
Bewältigungsprogramm, das grundsätzlich in jeder Belastungssituation aktiviert wird (Elenkov,
2006). Schon das morgendliche Aufstehen mit Änderung der Körperposition oder die körperliche
Anstrengung des Treppensteigens oder das Frühstück, alle Aktionen beanspruchen das
Stressprogramm, dessen Ablauf natürlich von Art und Dauer des Stressors abhängig ist.
Stressoren wirken über mentale, psychische, sensorische, visuelle, vegetative oder chemische
Signale unmittelbar zentral oder peripher über afferente Signale auf das Gehirn. Es kommt
simultan oder sequentiell zur Aktivierung des X
endokrinen Schaltzentrums, CRH (
Corticotropin-
Releasing Homon) aus dem Hypothalamus, und des Y
zentralnervösen Schaltzentrums, des
Locus coeruleus, der fast ausschließlich (90%) aus noradrenergen Neuronen besteht, in denen Noradrenalin als Signalgeber (Neurotransmitter) fungiert.
Noradrenalin und das hypothalamische
CRH stehen an der Spitze der Stress-Reaktionskette. Beide Hirnareale sind intensiv vernetzt. Noradrenalin verstärkt die CRH-Sekretion über adrenerge α1-Rezeptoren an den paraventrikulären Kernen des Hypothalamus, umgekehrt aktiviert CRH über CRH1-Rezeptoren
die Noradreanlinausschüttung aus dem Locus coeruleus. Dieser zentralen Reaktionsachse zugeordnet sind weitere CRH-Neurone der Medulla, AVP-Neurone (Vasopressin) des hypothalamischen PVN (
paraventrikulärer Nucleus) und gemischte CRH/AVP-Neurone. AVP wirkt synergistisch mit CRH. Das Neuropeptid Y hemmt Noradrenalin, stimuliert dagegen CRH, während Substanz P umgekehrt CRH hemmt und Noradrenalin stimuliert. Von den beiden zentralen Schaltzentren, PVN und LC, ausgehend werden die peripheren Reaktionszentren aktiviert: über CRH die Stresshormonachse mit ACTH aus der Hypophyse und über letzteres Cortisol und in begrenztem Umfang DHEA/S aus der Nebennierenrinde (NNR). Außerdem das Z
periphere autonome Nervensystem (ANS), Sympathikus und das Nebennierenmark (
Adrenalin;
SNS: Sympathoadrenomedulläres System) über Noradrenalin. ACTH stimuliert zusätzlich die LC-Aktivität durch Aktivierung von Schlüsselenzymen der Noradrenalinsynthese. Die Aktivierung der peripheren Komponenten des Stresssystems resultiert im systemischen Anstieg von Cortisol und den Katecholaminen. Cortisol hemmt schließlich im Feedback die zentrale ACTH-, CRH- und auch die zentrale Noradrenalinsekretion. Der Anstieg der Katecholamine (Noradrenalin, Adrenalin) die Aktivität des Schlüsselenzyms ihrer Synthese, der Tyrosinhydroxylase. Umgekehrt kann natürlich auch primär peripher über das autonome Nervensystem (Sympathikus, Parasympathikus) die Stressbewältigung in Gang gesetzt werden. Schließlich wirkt auch das [
Immunsystem maßgeblich am Zustandekommen der Stressreaktion mit und bildet neben der Stresshormonachse und dem zentralen und peripheren autonomen Nervensystem das vierte Reaktionszentrum der Stressbewältigung. Essentieller Bestandteil jeder akuten Stressreaktion, sei es psychischer, mentaler oder physischer Stress, körperliche Arbeit, Sport, Nahrungsaufnahme, Bedrohungssituationen oder Schlafmangel, ist eine zeitlich begrenzte zentrale Entzündungsreaktion mit Ausschüttung von proinflammatorischen Zytokinen wie TNF-alpha, Interleukin-1ß und IL-6 oder induzierbaren Entzündungsenzymen wie Cyclooxygenase 2 (COX2) bzw. der Stickoxidsynthetase iNOS, vor allem aus Gliazellen, über Noradrenalin. Die Entzündungsmediatoren stimulieren synergistisch die Ausschüttung von CRH, ACTH und Noradrenalin (Refojo, 2001). Vor allem IL-1ß werden ausgeprägte zentrale Wirkungen zugeschrieben. Umgekehrt führt auch periphere Immunaktivierung bei Infektionen, Verletzungen oder Entzündungen über die Ausschüttung der proinflammatorischen Zytokine zur zentralen Aktivierung. Die Zytokine aus der Peripherie können in begrenztem Maße die Bluthirnschranke überwinden und an zentrale hypothalamische und neuronale Zytokinrezeptoren binden. Auch Entzündungszellen können aus der Peripherie ins zentrale Nervensystem einwandern und den zentralen Entzündungsprozess unterhalten, sodass vor allem bei chronischen Entzündungen erhebliche zentrale Folgeerscheinungen resultieren.
HPT = Hypothalamus
Hypophysenvorderlappen
Nebennierenrinde
ZNS = Zentrales Nervensystem
ANS = Autonomes Nervensystem
IRS = immunresponse-System
STRESSHORMONACHSE
CRH: Die Cortisolsekretion wird durch CRH (
Corticotropin Releasing Hormone) aus dem
paraventrikulären Kernbereich des Hypothalamus (HPT) aktiviert, das an CRH1-Rezeptoren des
Hypophysenvorderlappens (HVL) andockt und ACTH stimuliert, das anschließend die Cortisolausschüttung aus der Nebennierenrinde anstößt. Die hormonelle Stress-Reaktionskette unterliegt enger Rückkopplung, da der Anstieg von Cortisol die CRH- und ACTH-Sekretion retrograd hemmt, bis es wieder zur Normalisierung der zirkulierenden Cortisolmenge kommt. Positives Feedback mit Verstärkung der CRH-Sekretion wird vor allem über Noradrenalin vermittelt, außerdem auch über Neuropeptid Y, Serotonin, Acetylcholin und durch Stress-induzierte
POMC-Peptide (
Propiomelanocorticotropine: ß-Endorphin, MSH) aus dem Nucleus arcuatus des Hypothalamus. Negatives Feedback dagegen durch autoregulatorische noradrenerge und CRH-Neurone über präsynaptische CRH1- bzw. α2-Rezeptoren, außerdem
durch GABA (
Gamma-Aminobuttersäure) und Substanz P, die in erster Linie über periphere Afferenzen aktiviert wird. CRH wirkt über CRH1- und CRH2-Rezeptoren, wobei die zentralen, neuronalen Wirkungen in
erster Linie über CRH1- und die peripheren neuronalen und hormonellen Effekte vorwiegend über
CRH2-Rezeptoren vermittelt werden. CRH1-Rezeptoren befinden sich außer in der Hypophyse
und dem Locus coeruleus auch im limbischen System, Vorderhirn, Kleinhirn und Rückenmark. CRH2-Rezeptoren vor allem im Gastrointestinaltrakt, an Hautzellen, Gefäßendothelien, in der
Skelett- und Herzmuskulatur sowie auch in einigen zentralen Regionen wie der Amygdala, dem lateralen Septum und dem Hypothalamus. Neben seinen zentralen Wirkungen (Hypophyse, LC, serotoninerges System) verfügt CRH über ausgeprägte periphere neuronale und hormonelle Funktionen (Dieterich, 1997). Sekretorisches, hormonaktives CRH ist an autonomen vegetativen Komponenten der Stressreaktion beteiligt, wirkt auf die Motivation und die Gemütslage, die Appetitsteuerung (anorektigene CRH-Wirkungen), die Energiebereitstellung und die Magen-Darmfunktion (intestinale Stressreaktion, Spasmen, Diarrhoen). Hervorzuheben sind auch die unmittelbaren CRH-Wirkungen auf Immunzellen (
immunCRH), die über CRH1-Rezeptoren vermittelt werden. Im Unterschied zu
Cortisol aktiviert immunCRH das zelluläre Immunsystem und steigert die Ausschüttung effektorischer und proinflammatorischer Zytokine aus T-Zellen und Monozyten/Makrophagen (TNF-alpha, IL-1ß, IL-6, IL-8). CRH und Substanz P zählen zu den potentesten Stimulantien von Mastzellen, steigern die Histaminsekretion, wirken vasodilatorisch und erhöhen die Gefäßpermeabilität. Direkt über CRH2-Rezeptoren in der Haut und indirekt über die Aktivierung
dermaler Mastzellen fördert Stress-induziertes CRH (und SP) die Exazerbation von entzündlichen und allergischen Hauterkankungen (Zoumakis, 2007). Die stressabhängige Aktivierung zentraler Mastzellen durch CRH (SP, NGF) hat weitgehende psychovegetative und kognitive Konsequenzen wie u.a. Auslösung von Angstreaktionen, emotionale Instabilität, kognitive Störungen. Die klinisch zu beobachtende Assoziation von Angsreaktionen mit Asthma, Nahrungsmittelallergien und Reizdarmsyndrom beruht auf diesen Mechanismen (Nautiyal, 2008). Über CRH2-Rezeptoren an Gefäßendothelien und der Herzmuskulatur stimuliert CRH die
Sekretion von proentzündlichen Faktoren, insbesondere die Bildung von Endothelin-1, das als der potenteste endogene Vasokonstriktor gilt. Zusätzlich wird die endotheliale Stickoxidsynthese (eNOS) gehemmt, was zur endothelialen Dysfunktion unter Stress beiträgt und eine Erklärung für die Entwicklung koronarer Komplikationen bei chronischem Stress und die massive Zunahme von Herzinfarkten bei Stresskranken liefert (Sheps 2002; Wilbert-Lampen, 2006). Eine weitere Gruppe von CRH-verwandten Stresshormonen, die Urocortine (Ucn I-III), wurde bisher weniger intensiv untersucht. Sie wirken in der Peripherie synergistisch mit CRH, ebenfalls über die CRH2-
Rezeptoren. CRH wirkt also einerseits als Neuropeptid und steuert die hormonelle Stressantwort, anderseits als peripheres Stresshormon, das auf direktem Wege eine Vielfalt vegetativer und immunologischer Reaktionen auslöst (Chrousos, 2000). Im Gegensatz zum HPT-HVL-NNR-Regelkreis unterliegen die extrahypothalamischen CRH-Wirkungen nicht der Cortisol-Feedbackregulation.
S bstanz P
poth lamus
Cortisol Das wichtigste periphere Stresshormon, das adrenale Cortisol wird aus Cholesterin über
Pregnenolon – Progesteron – 17-OH-Progesteron und 11-Desoxicortisol gebildet. Es hat ein
umfassendes Wirkungsspektrum, das die optimale Anpassung des Organismus an akute und
wiederkehrende Belastungssituationen maßgeblich unterstützt: Stoffwechselaktivierung,
Energiebereitstellung durch Glykogenolyse, Glukoneogenese aus Aminosäuren, allerdings auf
Kosten von Muskeleiweiß, Fettumverteilung, Temperatursteigerung, hormonelle Umstellung,
emotionale und kognitive Aktivierung, Schmerzhemmung, Entzündungshemmung. Zentral
blockiert Cortisol die Sekretion von Wachstumshormon, Gonadotropinen und TSH, peripher wirkt
Cortisol vor allem immunsuppressiv, hemmt die unspezifische Entzündungsantwort und hemmt
die zelluläre Immunabwehr.
Cortisol diffundiert auf Grund seines lipophilen Charakters frei durch die Oberflächenmembran
von Zielzellen und bindet intrazellulär spezifisch an zytoplasmatische Glukocorticoidrezeptoren
(GR), mit geringerer Affinität auch an Mineralocorticoidrezeptoren (MR). Die GC-Rezeptoren
kommen in zahlreichen, strukturell und funktionell unterschiedlichen Varianten vor, die für die
vielfältigen Wirkungen von Cortisol verantwortlich sind. Durch Bindung an zelluläre Chaperone, in
erster Linie das Hitze-Schock-Protein HSP90, sind die GC-Rezeptoren im Zytplasma der
Zielzellen präformiert. Nach Bindung von Cortisol entsteht ein beweglicher GR-Komplex, der in
den Zellkern einwandert und dort Cortisol-sensitive Gene aktiviert. Zu den wichtigsten
Eigenschaften der GRs gehört ihre direkte Interaktion mit dem zentralen Entzündungsschalter
der Zellen, dem Redox-sensitiven NF-kB Tranferfaktor. Das dynamische Gleichgewicht zwischen
GR- und NFkB-Aktivität entscheidet maßgeblich über das Ausmaß entzündlicher Aktivität. Je
höher der Cortisolspiegel und je höher der stressabhängige Anstieg desto geringer die zelluläre
Enzündungsaktivität. Chronischer Stress vermindert jedoch die Fähigkeit von Cortisol, NF-kB
inaktiv zu halten und die Produktion inflammatorischer Zytokine zu blockieren (Miller, 2008),
sodass auch bei stressbedingtem, chronischem Hypercortisolismus die Entzündungsaktivität im
Organismus ansteigen kann. Deren Ausmaß hängt nicht zuletzt von pro- und anitentzündlichen Reaktionen ab, die durch peripheres
immunCRH bestimmt werden. Bei Zuständen mit Verminderung der HVL-NNR-Aktivität (CFS, FMS, PTSD, Burnout) ist konsequenterweise die Entzündungsaktivität häufig erhöht, bei der schweren Depression kommt es jedoch parallel zur CRH- und Cortisol-Hyperaktivität ebenfalls zu erhöhter entzündlicher Aktivität. Die Cortisolproduktion unterliegt einem strengen 24h-Rhythmus. Die Hauptproduktion findet in der zweiten Nachthälfte mit einem frühmorgendlichen Maximum statt, sodass zum Tagesbeginn die optimale Cortisolmenge zu Verfügung steht. Kurz nach dem Aufstehen kommt es allerdings noch zu einem weiteren, kurzfristigen Cortisolanstieg (30 – 60 min), dem sogenannten CAR (
Cortisol Awakening Response). Der CAR kommt nicht allein durch die Aktivierung der Stressachse nach dem Aufstehen zustande, die Mitwirkung weiterer Gehirnstrukturen wie vor allem des Hippocampus gilt als gesichert (Fries, 2008). Der CAR wird als guter Parameter für den allgemeinen Gesundheitszustand und die Stressbelastung bzw. Stressresistenz angesehen. Nach dem morgendlichen Peak fällt Cortisol bereits bis zum Mittag rasch ab, um im weiteren Tagesverlauf bis auf ein nächtliches Minimum abzusinken. Dieser physiologische Tagesrhythmus wird bei Auseinandersetzung mit Stressoren durch kurzfristige Anstiege überlagert. Mit zunehmendem Alter flacht die Tageskurve bei unveränderter Gesamtproduktion ab. Allerdings ist im Alter – ähnlich Untrainierten - die Stressempfindlichkeit erhöht, in Belastungssituationen steigt Cortisol häufig stärker an. Training verbessert die Stressresistenz, der Ruhe-Cortisolspiegel sinkt ab, der Anstieg unter Belastung ist geringer. Die hormonaktive Cortisolkonzentration bei Frauen ist niedriger als bei Männern, allerdings liegt die Konzentration von Gesamtcortisol im Serum bei Frauen höher, da Östrogen-abhängig ebenso wie SHBG (
Sexualhormon-bindendes Globulin) auch mehr Cortisol-bindendes Globulin (
CBG) vorhanden ist. Als lipophiles, nicht-wasserlösliches Molekül liegt Cortisol im Serum weit überwiegend anTrägereiweiße gebunden vor (97-98% Bindung an CBG und Albumin), nur ein geringer Teil von 1-3% ist ungebunden, biologisch aktiv verfügbar. Während die Bestimmung des Gesamtcortisols im Serum oder im 24h-Urin für die Differentialdiagnose organischer Störungen der HPT-HVL-NNR-Achse geeignet ist, ist für funktionelle Analysen der Cortisolbestimmung im Speichel der Vorzug zu geben. Speichelcortisol repräsentiert den aktiven, biologisch frei verfügbaren Anteil des zirkulierenden Gesamthormons, der wegen seines lipophilen Charakters die Endothelien der Speicheldrüsen ungehindert durchwandern kann. Die Konzentration des Speichelcortisols liegt dementsprechend nur bei 1 – 9% der Serumkonzentration. Die freie, ungebundene Hormonfraktion im Speichel entspricht eher der momentanen Hormonaktivität, während die Gesamtkonzentration im Serum eher den Reservepool des Hormons repräsentiert. Die Relation von freiem zu Gesamthormon im Serum unterliegt Schwankungen, abhängig vom momentanen Sekretionsstatus und von der Trägerproteinkonzentration bzw. -sättigung. Cortisol wird nach der Sekretion frei ins Blut sezerniert und anschließend an Trägerproteine gebunden, sodass der freie Anteil in aktiven sekretorischen Phasen höher als in Ruhephasen ist. Am Morgen, in der Hauptsekretionsphase, sind die Speichelwerte daher relativ höher, im Tagesverlauf dagegen niedriger als die Serumwerte, was einen steileren Gradienten bedingt. Speichelwerte können in Sekretionsintervallen (falsch) niedrig, scheinbar zu niedrig, sein während Serumbestimmungen ein stabileres Bild der hormonellen Situation liefern. Die Serumwerte sind stark durch die vorhandene Trägerproteinmenge beeinflusst, deren hepatische Syntheserate östrogenabhängig ist. Bei akuten Belastungen steigt die morgendliche Cortisolsekretion an, bei chronischem Stress ist die Cortisol-Tageskurve insgesamt zu höheren Konzentrationen verschoben, bis es bei anhaltender Überlastung schließlich zu partiellem oder totalem Ausfall der Tagesrhythmik kommt (Björntorp, 2000; Hellhammer, 2008). Die nächtliche Cortisolproduktion wird kaum noch oder nicht mehr aktiviert, im Tagesverlauf bleibt die NNR-Aktivität niedrig oder reagiert nur auf aktuelle Belastungssituationen. Die Cortisol-Tageskurve ist daher ein direktes, zeitnahes Abbild der individuellen Belastungsituation.
Cortisol wirkt zentral prooxidativ, hemmt die Neurogenese u.a. durch Hemmung des
Neurogenesefaktors BDNF (
Brain-derived neurotrophic Factor) und begünstigt die Apoptose, den
Untergang von Nervenzellen. Während kurzfristige Cortisolanstiege in akuten
Belastungssituationen in der Regel keine Langzeitfolgen haben, wirkt anhaltender
Cortisolüberschuss dagegen neuro- und zytotoxisch und führt zu bleibenden funktionellen und
morphologischen Veränderungen einzelner Hirnregionen. Je höher die mit chronischem Stress
oder bestimmten psychischen Erkrankungen verbundene entzündliche Aktivität infolge
verminderter Cortisolwirksamkeit desto geringer das Hippocampusvolumen (Marsland, 2008)
Extremer kurzfristiger Stress (PTSD) kann allerdings zu ähnlichen Langzeitfolgen wie chronischer
Stress führen.
DHEA
Ausgangssubstanz der Mehrzahl der adrenalen Steroidhormone ist Dehydroepiandrosteron
(DHEA), das aus Cholesterin über Pregnenolon – 17-OH-Pregnenolon in der Nebennierenrinde
gebildet wird und wie Cortisol über die HPT-HVL-Achse mit ACTH reguliert wird. Allerdings fehlt
die für Cortisol charakteristische enge Rückkopplung. DHEA wird zum Teil bereits in der NNR
oder später in peripheren Organen zum wasserlöslichen DHEAS sulfatiert, das wegen seines
hydrophilen Charakters im Unterschied zu den übrigen Steroidhormonen ohne Bindung an ein
Carrierprotein frei im Blut zirkulieren kann. DHEA/DHEAS wird in geringer Menge auch
extraadrenal gebildet. Im Gehirn wird DHEA lokal als Neurohormon von Astrozyten und
Neuronen synthetisiert, außerdem aus der Peripherie über die Blut-Hirnschranke angereichert.
Die DHEA-Konzentation im Gehirn liegt bis zu 10fach, im Durchschnitt 6,9fach höher als im Blut
(Maninger, 2008). In der Peripherie dient DHEA in erster Linie als Vorstufe der Sexualhormone,
hat aber offensichtlich auch eigene hormonelle Effekte. Die Tagesrhythmik von DHEA/S ist
weniger ausgeprägt als bei Cortisol, der Konzentrationsunterschied zwischen Morgen- und
Abendwert liegt jedoch auch für DHEAS beim 2 – 3 fachen, für freies DHEA kann er sogar bis
zum 10 fachen betragen. Die maximale Serumkonzentration wird im frühen Erwachsenenalter
(20 – 30 J) erreicht und fällt anschließend bis zum 70. LJ nahezu linear um bis zu 90% ab. Außer
im Alter ist DHEA auch bei chronischem Stress oder Autoimmunerkrankungen vermindert.
In vieler Hinsicht wirkt DHEA/S Cortisol-antagonistisch. Es hat leicht anabole/androgene Effekte,
fördert den Muskelaufbau und den Fettabbau, wirkt mäßig lipidsenkend und erhöht HDL-
Cholesterin. Wie Cortisol wirkt DHEA/S antientzündlich, wobei vor allem sein antioxidativer Effekt
mit Hemmung der ROS-Bildung und konsekutiver Hemmung der NF-kB-Aktivierung zum Tragen
kommt. im Unterschied zu Cortisol unterstützt DHEA die zelluläre Immunabwehr und stärkt die
TH1-Immunachse. Das Plasmaverhältnis von Cortisol zu DHEAS, unabhängig von den absoluten
Konzentrationen, gilt als Ausdruck der „anabolen Balance".
Neurohormonelles DHEA/S wirkt vorwiegend als Neuromodulator (Perez-Neri, 2008). Es
moduliert die Synthese und Rezeptoraktiviät von Glutamat und GABA, zusätzlich auch die
Serotonin- und Dopaminwirkung und die Aktivität der Stickoxidsynthetase nNOS. Es wirkt
außerdem auf die Neurogenese, die Apoptose von Nervenzellen, die frühe Gehirnentwicklung
und die Kognition. DHEA und DHEAS stimulieren die Dopaminsekretion und erhöhen die
extraneuronale Dopaminaktivität. In hoher Dosis hat DHEA daher psychostimulierende Effekte
und kann im Extremfall manisches Verhalten begünstigen. DHEA und DHEAS stimulieren vor
allem die Sekretion von Glutamat und die Aktivität des NMDA/Glutamatrezeptors im
Hippocampus und steigern die Neurogenese, wobei sie Cortisol-antagonistisch wirken. Der
DHEAS/Cortisol-Quotient korreliert positiv mit der Neurogeneserate im Hippocampus. Wegen
seines Glutamat- und Dopamin-fördernden Effekts hat DHEA in hoher Dosis günstige Wirkungen
bei der Schizophrenie gezeigt.
Ähnlich Estradiol stimuliert DHEA (nicht DHEAS) die Synthese von Stickoxid und fördert die
Hirndurchblutung und Kognition. Schließlich moduliert DHEA das Serotoninsystem, hemmt den
5HT-Abbau und steigert die 5HT-Rezeptorwirkung, was seine antidepressive Wirksamkeit und
seinen synergistischen Effekt mitAntidepressiva erklärt. Im GABA-System wirkt DHEA
überwiegend hemmend, verstärkt jedoch zum Teil auch GABA-Effekte. Orale DHEA-Gabe wirkt
im Unterschied zu Estradiol angstlösend.
Insgesamt wirkt DHEA/S im Gehirn neuroprotektiv, stimuliert die Neurogeneseund das
Wachstum von Neuriten und hemmt gleichzeitig den Untergang von Nervenzellen (Apoptose). Es
wirkt leicht antioxidativ, bremst die Amyloidaggregation (Alzheimer) und hemmt die
Glucocortikoidtoxizität. Als Neuromodulator fördert DHEA die Motivation, wirkt
stimmungsaufhellend, angstlösend und antidepressiv und steigert die kognitive
Leistungsfähigkeit. Verschiedene psychiatrische Erkrankungen sind mit niedrigem DHEA bzw.
erniedrigtem DHEA/Cortisol-Quotienten assoziiert: Depressionen, Angststörungen, PTSD,
Schizophrenie und Demenz (Manninger, 2008).
NEURONALE STRESSACHSE, NEUROTRANSMITTER
Die neuronale Signalübermittlung von Nerv zu Nerv, von Nerv zu Ganglion, von efferenten
Nerven zum peripherem Erfolgsorgan (Muskel, sensorische Organe, Magen-Darmtrakt, etc.) bzw.
von der Peripherie über afferente Nervenbahnen zum ZNS erfolgt über spezifische Signalstoffe
der Nervenzellen, die Neurotransmitter, die an spezifische potsynaptische Rezeptoren der
Erfolgsorgane binden. Die Neurotransmitter sind entweder selbst Aminosäuren, biogene Amine,
die sich von Aminosäuren ableiten, Neuropeptide oder atypische Signalstoffe wie z.B. Stickoxid
(NO).
Die wichtigsten, die klassischen Neurotransmitter leiten sich von Aminosäuren ab, Serotonin aus
Tryptophan und die drei Katecholamine Dopamin, Noradrenalin und Adrenalin aus Tyrosin bzw.
Phenylalanin. Sie sind sowohl zentral als auch peripher aktiv. Die vorwiegend zentral aktiven
Neurotransmitter GABA, Glycin und Glutamat sind ihrerseits Aminosäuren. Sie sind in erster Linie
interneuronal mit neuromodulierender Funktion wirksam. Alle Neurotransmitter werden in den
entsprechend spezialisierten Nervenzellen (Neurone, die die über die für die Synthese des
betreffenden Neurotransmitters erforderlichen Enzyme verfügen) gebildet bzw. angereichert und
in speziellen Vesikeln gespeichert.
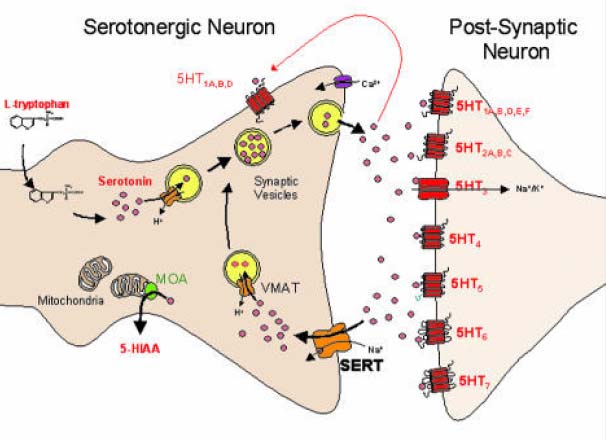
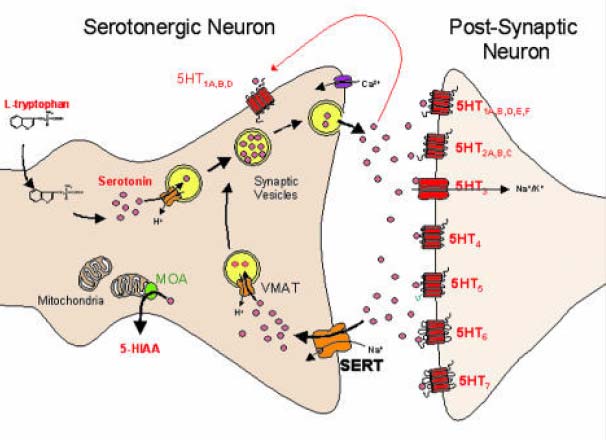
Nach Stimulation des Nerven und elektrischer Übermittlung des Aktivierungssignals zum synaptischen Nervenende wandern die Neurotransmittervesikel zur präsynaptischen Zellmembran und geben Calcium-abhängig die gespeicherten Neurotransmitter in den synaptischen Spalt ab, wo sie an spezifische Rezeptoren der postsynaptischen Membran binden und exzitatorische oder inhibitorische Signale an die Erfolgsorgane übermitteln. Die sezernierten Neurotransmitter werden umgehend aus dem synaptischen Spalt entfernt, wobei der Reuptake (Wiederaufnahme) in das präsynaptische Nervenende mit Abstand am wichtigsten ist und die laterale Diffusion nachrangig ist. Die resorbierten Neurotransmitter werden zum Teil wieder in Vesikel verpackt, wo sie für die erneute Signalübermittlung bereitstehen, oder sie werden im präsynaptischen Ende enzymatisch metabolisiert. Der Reuptake erfolgt über spezifische Transportermoleküle wie den SERT (Serotonin-Reuptake-Transporter) oder NET bzw. DAT1, die Transporter für Noradrenalin und Dopamin. Diese Transporter sind in identischer Form in zahlreichen spezialisierten Zellen zu finden, z.B. für den Uptake von Serotonin in Kapillarendothelien der BHS, die Aufnahme aus dem Gastrointestinum und den Uptake aus dem Serum in Thrombozyten und andere Blutzellen, sowie den Reuptake in Nierenzellen. Der Nervenimpuls wird durch Bindung sezernierter Neurotransmittermoleküle an präsynaptische, inhibitorische Rezeptoren terminiert.
Abbildung: Neuronale Synapse am Beispiel
eines serotoninergen Neurons.
Der Neurotransmitter (Serotonin = 5HT) wird
aus Tryptophan gebildet,
in Versikeln gespeichert und nach Exzitation
und Ca-Einstrom in den synaptischen Spalt
abgegeben, wo er an postsynaptische 5HT-
(5HT1N-7N) bindet. Übeschüssiger
Neurotransmitter (NT) wird über den Reup-
take-Transporter SERT (oder NET) wieder
in das synaptische Nervenende rücktrans-
portiert. Er wird anschließend entweder über
einen SERT-analogen vesikulären Trans-
porter (VMAT = versikulärer Monoamintrans-
porter) in nascente NT-Vesikel aufgenommen,
oder über Monoaminoxidase A (MOA)
zu 5-HIAA (5-Hydroxyindolessigsäure) meta-
bolisiert und abgegeben.
Blut-Hirn-Schranke
Die Blut-HirnSchrank (BHS) ist eine weitestgehend undurchlässige Zellschicht zwischen
Blutkapillaren und Liquorraum. Die polar gebauten Neurotransmitter (NT) können generell nicht
über die Blut-Hirn-Schranke (BHS) ins Gehirn gelangen, sodass das ZNS nicht durch Signalstoffe
aus der Peripherie überschwemmt werden kann. Sie werden im Gegenteil aktiv durch die mit den
synaptischen Reuptake-Transportern identischen Carrier aus dem ZNS eliminiert (SERT, NET,
DAT1, GAT2). Für ihre Aminosäure-Vorstufen existieren spezifische Carriersysteme, sowohl auf
der luminalen (Blut-Endothelgrenze) als auch auf der abluminalen Seite (Liquor-
Endothelgrenzfläche), die zum Teil identisch sind. Eine Ausnahme bildet Glutamin, die Vorstufe
der beiden mengenmäßig dominanten Neurotransmitter, GABA und Glutamat, das in wesentlich
höherer Konzentration im Gehirn vorkommt, dessen Konzentration jedoch ebenfalls durch einen
abluminalen Carrier begrenzt wird (LAT/System N). Für den Transport und Abtransport des
potentiell neurotoxischen Glutamat aus dem ZNS in die Peripherie exisiterien gleich 3 spezifische
Carriersysteme, EAAT1-3. Glutamat wird 1:1 molar im Austausch gegen Cystin transportiert, das
essentiell für die Synthese des herausragend wichtigen Antioxidans und Detox-Agens Glutathion ist. Auch Taurin nimmt wegen seiner neuroprotektiven, neuromodulatorischen Bedeutung im ZNS eine Sonderstellung ein, sowohl luminal als auch abluminal ist ein eigenes Transportsystem (TAUT) vorhanden (Ohtsuki, 2004).
Für verschiedene Peptide, Proteine und andere polare Stoffe existieren weitere spezifische
Transportersysteme, u.a. für Insulin, AVP, Enkephalin, DHEAS, etc. Auch für proentzündliche
Zytokine sind spezifische Carier vorhanden, die Entzündungssignale aus der Peripherie ins ZNS
weiterleiten. Für die Energieversorgung des Gehirns, die auf Glukose basiert, ist ebenfalls ein
spezifisches Carriersystem mit hoher Kapazität (GLUT1) vorhanden. Auch für Creatin, das im
Gehirn besondere Bedeutung als Intermediär-Energiespeicher hat und 180fach höher
konzentriert ist als im Blut, exisitiert ein spezifischer Transporter (CRA).
Katecholamine
Das Pendant zu CRH auf der hormonellen Seite ist Noradrenalin, der wichtigste Neurotransmitter
der zentralen und autonomen neuronalen Stressantwort. Wie bereit erwähnt steht der Locus
coeruleus als noradrenerges Zentrum in enger Kooperation mit dem Hypothalamus (CRH) an der
Spitze der Stress-Reaktionskaskade. CRH aktiviert Nordrenalin und umgekehrt Noradrenalin
CRH. Noradrenalin stimuliert außerdem im Verlauf der akuten Stressreaktion nachgeordnete
dopaminerge und serotoninerge Hirnzentren. Noradrenerge, efferente Nerven aktivieren das
periphere sympathische Nervensystem und die Nebenniere, sowohl die Nebennierenrinde (NNR)
mit Steigerung dder Cortisolsekretion als auch das Nebennierenmark (NNM), woraufhin
Adrenalin, das kardiovaskulär und metabolisch maßgebliche Neurohormon, ausgeschüttet wird.
Noradrenalin ist der Initiator der schnellen Stressadaptation, der Motor der „Fight or Flight"-
Reaktion.
Noradrenalin zählt mit Adrenalin und Dopamin zu den Katecholaminen, die aus der Aminosäure
Tyrosin bzw. Phenylalanin synthetisiert werden, wobei Vitamin C, Vitamin B6, Kupfer,
Magnesium und Folat (Tetrahydrobiopterin) essentielle Kofaktoren der an deren Synthese
beteiligten Enzyme sind. Das limitierende Enzym der Katecholaminsynthese ist die
Tyrosinhydroxylase, die den Schritt von Tyrosin zu DOPA katalysiert. Sie wird durch die
Katecholamine selbst (negatives Feedback durch Dopamin und Noradrenalin) und durch
Glukocorticoide (Cortisol) gehemmt.
Vitamin B6 (Pyridoxalphosphat) ist von essentieller Bedeutung für die Synthese der
Katecholamine, es fungiert als Kofaktor von zwei enzymatischen Schritten auf dem Weg zum
Adrenalin.Auch bei der Synthese des Neurotransmitters PEA (Phenylethylamin), der
ausschließlich aus Phenylalanin gebildet wird und partiell als Kotransmitter von Noradrenalin
fungiert, ist Pyridoxalphosphat beteiligt.
a renalin
sen, upfe
opa in-ß-
r A- Decarb
car oxylase
Reize, NG
Zusammen mit Folsäure, Vitamin B2 und B12 ist Vitamin B6 auch in den Metabolismus
schwefelhaltiger Aminosäuren (Methionin, Cystein) involviert und generell für die Bereitstellung
von Methylgruppen mitverantwortlich. Defizite durch geringes Angebot oder erhöhten Verbrauch
eines oder mehrerer dieser Vitamine können ebenso wie Methionin- bzw. Betainmangel (Betain =
Trimethylglycin) zur Homocysteinämie und Mangel an S-Adenosylmethionin (SAMe) führen.
SAMe ist der zentrale Methylgruppendonator des Organismus, der für mehr als 80
Methylierungsreaktionen verantwortlich ist, der auch die Methylgruppe für die Umwandlung von
Noradrenalin zu Adrenalin liefert. Vitamin C ist wichtig für die Synthese von Noradrenalin. Auch
an der Bildung von Cholin, der Vorstufe von Acetylcholin, des Neurotransmitters des
parasympathischen autonomen Nervensystems, und dem Baustein der Membranphospholipide
ist SAMe beteiligt.
Dopamin
wurde um 1910 entdeckt und lange nur als Vorstufe der beiden anderen Katecholamine,
Noradrenalin und Adrenalin, angesehen. Erst später wurde offenbar, dass Dopamin (DA) nicht
nur als Noradrenalinpräkursor dient, sondern selbst zu den wichtigsten Neurotransmittern
(Botenstoffe) im ZNS gehört, und dass in verschiedenen Hirnregionen (Substantia nigra,
Striatum), die gar kein Noradrenalin enthalten, Dopamin als primärer Neurotransmitter fungiert.
Dopaminerge Kerne im Nucleus arcuatus des Hypothalamus innervieren die Hypophyse, wo DA
u.a. die Prolaktinsekretion limitiert. Außerdem ist das „mesostriatale dopaminerge System", das
limbische Strukturen innerviert und Sensorik, Emotionen und Affekt beeinflusst, hervorzuheben.
Dopaminerge Neurone sind häufig überaus intensiv über Dendriten mit anderen neuronalen
Strukturen vernetzt und wirken stark neuromodulatorisch. Ein herausragendes Beispiel ist das
„nigrostriatale" System (Neuronengeflecht Substanzia nigra – Striatum), in dem einzelne
Dopaminneurone bis zu 100.000 Dendriten ausbilden. Seit 1989 sind auch efferente, periphere
dopaminerge Neurone nachgewiesen.
Dopaminerge Kerne des Gehirns werden über Noradrenalin und CRH stimuliert. Im
Zusammenspiel mit Noradrenalin ist DA die zentrale stimulierende (exzitatorische) Kraft. Es kann
als der „Kraftstoff" des Gehirns bezeichnet werden, der den Organismus antreibt. DA steuert
maßgeblich Motorik, Koordination, Konzentration und geistige Wachheit. Zusammen mit
Serotonin und Noradrenalin wirkt es stimmungsaufhellend und motivierend, sodass heute neben
Serotonin- und Noradrenalin-verstärkenden vermehrt auch Dopaminpräparate in der Behandlung
von Depressionen zum Einsatz kommen.
Im Alter geht die DA-Syntheseleistung des ZNS zurück, auch die Anzahl der Dopaminrezeptoren
sinkt ab, sodass die Gefahr des symptomatischen Dopaminmangels zunimmt. Morbus Parkinson,
bei dessen Entstehung Alter, genetische Disposition und zusätzliche toxische Umweltfaktoren
zusammenwirken, ist die Extremform des Dopamindefizits mit verminderter Synthesekapazität im
Striatum infoge Schädigung der Substantia nigra (Nigrostriatales System) und mit verminderter
Dopaminrezeptordichte (D2-Rezeptoren).
DA wirkt über prä- und postsynaptische Rezeptoren. Die postsynaptischen Dopaminrezeptoren
gliedern sich in zwei Hauptfamilien: die D1-Familie mit D1- und D5-Rezeptoren und die D2-
Familie mit D2-, D3- und D4-Rezeptoren. DA wird wie alle anderen Neurotransmitter nach der
Freisetzung entweder in das präsynaptische Nervenende zurücktransportiert oder metabolisiert.
Bindung von DA an präsynaptische Rezeptoren beendet autoregulativ die Dopaminsekretion. Für
den Abbau stehen zwei Wege zu Verfügung: einmal über die COMT (Catechol-O-
Methyltransferase) zu 3-Methoxytryptamin und weiter über die MAO (Monoaminoxidase) bis zur
Homovanillinsäure (HVA); oder zuerst über die MAO zu DOPAC und dann über die COMT zur
HVA, deren Konzentration doppelt so hoch ist wie die der VMS, der Nor/Adrenalinendstufe.
Ein erheblicher Anteil der Dopaminsynthese entfällt auf den Intestinaltrakt (45%; Eisenhofer
2004).
Noradrenalin
wurde um 1940 als eigenständiger Neurotransmitter identifiziert. Es ist, wie bereits mehrfach
betont, das wichtigste Katecholamin im ZNS, ebenso der dominate Neurotransmitter des
peripheren sympathischen Nervensystems (vor Adrenalin). Der Locus coeruleus (LC) stellt die
Hauptquelle für NA im Gehirn dar, ca. 20% der LC-Neurone exprimieren gleichzeitig das eher
inhibitorische wirksame Neuropeptid Y (NPY), 70% gleichzeitig Galanin, ein Neuropeptid, das
ebenfalls überwiegend inhibitorisch wirkt mit Ähnlichkeit zu GABA. Daneben dominiert NA im L.
subcoeruleus, der Formatio reticularis und dem Tractus solitarius. NA aus dem LC aktiviert nicht
nur die hypothalamische CRH-Sekretion sondern wirkt auch auf Thalamus, Hippocampus,
Amygdala, Septum und Rückenmark.
Noradrenalin wird aus Dopamin über die Vitamin C-abhängige Dopamin-ß-Hydroxylase gebildet.
Das limitierende Enzym der NA-Synthese ist die in den ersten Schritt der Dopaminsynthese
involvierte Tyrosinhydroxylase (Th), die durch Dopamin- und durch Noradrenalinüberschuss
autoregulativ gehemmt wird. Die Th besitzt strukturell weitgehende Homologie mit der Dopamin-
ß-Hydroxylase, beide werden durch Dopamin- oder NA-Mangel aktiviert. Zentrales oder
sympathogenes Noradrenalin wird nach synaptischem Release durch einen für alle
Katecholamine identischen Reuptake-Transporter (NET) wieder in das noradrenerge synaptische
Terminal aufgenommen, zum Teil wieder in nascente sekretorische Vesikel eingebaut oder
metabolisiert.
Die Metabolisierung verläuft ausschließlich über die mitochondriale MAO mit Deaminierung zu
DHPG, das nach Abgabe in die Blutbahn in der Leber über die COMT methyliert und weiter bis
zur Vanillinmandelsäure (VMS) abgebaut wird. Adrenomedulläres Noradrenalin und Adrenalin
werden dagegen über die zytoplasmatische adrenale COMT zu Nor/Metanephin (NMN bzw. NM)
methyliert und anschließend über adrenale MAO zu MHPG umgewandelt. NMN, MN und MHPG
werden nach Abgabe ins Blut in der Leber weiter zur VMS abgebaut.
NA bindet an postsynaptische α- und ß-Rezeptoren. Es erhöht das Aufmerksamkeits-
/Wachheitsniveau, fördert Konzentration, Motivation und Motorik. Kurzfristig wirkt NA
proentzündlich über die Aktivierung von NF-kB, wobei vor allem IL-6 und die anschließende
hepatische CRP-Produktion massiv stimuliert werden (Marz, 1998; Bethin, 2000). Langfristig
hemmt NA analog Cortisol die zelluläre Immunfunktion, begünstigt den TH1�TH2-Switch und
blockiert die Synthese des proentzündlichen Leitzytokins TNF-alpha. NA-Mangel führt zu
Motivationsabfall, Antriebs- und Konzentrationsschwäche, kognitiven Einbußen mit Störung des
Kurzzeitgedächtnis und Stimmungsabfall bis zu Depressionen.
Adrenalin
wurde bereits1856 von Vulpian und Bates in der Nebenniere entdeckt und 1904 in seiner
chemischen Struktur beschrieben. Adrenalin (Epinephrin) wird vorrangig im Nebennierenmark
aus Dopamin über Noradrenalin synthetisiert, wobei die erforderliche Methylgruppe über SAMe
(S-Adenosylmethionin) beigesteuert wird. Cortisol stimuliert die Umwandlung von Noradrenalin in
Adrenalin. Es wird wie die neuronalen Katecholamine in Vesikeln gespeichert und
zytoplasmatisch rezirkuliert. Das Verhältnis von Adrenalin zu Noradrenalin liegt in der Nebenniere
bei ca. 4:1. Adrenalin hat in erster Linie hormonelle Funktionen, als Neurotransmitter spielt es
eine untergeordnete Rolle, da im ZNS kaum adrenerge Neurone vorkommen und auch im
peripheren Sympathikussystem Noradrenalin dominiert.
Adrenalin ist ein Stresshormon, das über Noradrenalin aktiviert wird. Seine Plasmahalbwertzeit
beträgt nur 1-3 min. Es steigert hormonell und neuronal über ß-Rezeptoren die Herz-
Kreislaufaktivität, die Pulsfrequenz, das Herzminutenvolumen, den Blutdruck und zentralisiert die
Durchblutung durch Kontraktion kleinerer Haut- und Nierengefäße über α1-Rezeptoren und
gleichzeitg Weiterstellung von zentralen und Muskelgefäßen über ß2-Rezeptoren. Es erhöht
außerdem analog zu Noradrenalin die mentale Aktivität, das Konzentrationsvermögen, Motivation
und Denkleistung und hemmt.die Magen-Darm-Tätigkeit. Außerdem ist Adrenalin als
Stresshormon von herausragender Bedeutung für die rasche Energiebereitstellung, es stimuliert
die Glykolyse/Gluconeogenese, die Lipolyse und steigert die Sauerstoffaufnahme und
Atemfrequenz.
Serotonin
Ist seit etwa 1960 als Neurotransmitter erkannt und zählt inzwischen unzweifelhaft zu den
wichtigsten zentralen Neurotransmittern. Es wird aus der Aminosäure Tryptophan über 5-
Hydroxytryptophan (5HTP) durch das Enzym Tryptophanhydroxylase (Tph) unter Mitwirkung von
Vitamin B6 gebildet. 5HTP wird über die gleiche Decarboxylase, über die DOPA in Dopamin
umgewandelt wird, zu Serotonin (5HT, 5-Hydroxytryptamin) decarboxyliert. Typtophan konkurriert
mit zahlreichen langkettigen Aminosäuren (Phenylalanin, Tyrosin, Leucin, Isoleucin, Threonin,
Methionin, Serin, Valin, Histidin) um den Carrier-abhängigen Transport (LAT1) über die Blut-Hirn-
Schranke ins Gehirn. Ein breites postprandiales Aminosäureangebot (Eiweißmahlzeit) wirkt
daher eher hinderlich für die Tryptophanaufnahme. Muskelarbeit und Insulin stimulieren die
Aufnahme der Aminosäuren, nicht jedoch von Tryptophan (Trp), in die Muskelzelle, da dieses sich durch Bindung an Albumin dem Muskeluptake entziehen kann (Newsholme, 2006). Daher verbessert Sport oder der Insulinanstieg nach einer kohlehydratreichen Ernährung das Trp-Angebot für das ZNS, während eiweißreiche Nahrung kompetitive Wirkung hat. Eine gängige Erklärung des bei Serotoninmangel charakteristischen Heißhungers auf Kohlehydrate („Craving"), der allerdings auch ein häufiges Zeichen von GABA-Mangel ist (s.u.). Die Tph ist das limitierende Enzym der 5HT-Synthese und wird wie die eng verwandte Tyrosinhydroxylase durch den betreffenden Neurotransmitter, in diesem Fall Serotonin, im Überschuss gehemmt. Sie kommt in zwei unterschiedlichen Formen vor, als Tph1 im Magen-Darmtrakt (MDT) und als Tph2 im Nervensystem (Walther, 2002). Die bei weitem größte Menge an Serotonin wird in den enterochromaffinen Zellen des MDT gebildet, wo es parakrin auf Motilität, Motorik und Resorptionsfunktion wirkt, unterstützt von serotoninergen, efferenten Nervenfasern, CRH-Neuronen und zahlreichen weiteren neuroendokrine Faktoren (VIP, Dopamin, Adrenalin, etc.). Überschüssiges 5HT wird aus dem MDT über den Serotonin-Transporter (SERT) resorbiert und gelangt über die Blutbahn zu den Thrombozyten, wo es über das gleiche SERT-System aufgenommen und gespeichert wird. Auch Monozyten und Lymphozyten speichern Serotonin.
Try tophan
-HT 5-Hyd
Generell ist bei Frauen die zentrale Serotonin-Synthesekapazität niedriger als bei Männern (Nishizawa, 1997). Vielfach untersucht sind genetische Veränderungen der Tph, die Tph1 und Tph2 unterschiedlich betreffen können, sodass z.B. im ZNS genetisch bedingte Veränderungen der Enzymaktivität bei normaler Tph1-Aktivität im MDT vorkommen können - und umgekehrt. Insgesamt sind Genvarianten der Tph mit eingeschränkter Enzymaktivität häufiger beschrieben. Im Extremfall kann die zentrale Serotoninsynthese um bis zu 80% vermindert sein. Serotoninerge Neurone sind über das ganze ZNS verteilt. Serotoninerge Synapsen fungieren grundsätzlich wie die anderen neuronalen Synapsen. Serotonin wird neuronal aus Tryptophan über 5HTP gebildet und in Vesikeln gespeichert. Aktivierungssignale führen zur Abgabe von Serotonin in den synaptischen Spalt und Bindung an postsynaptische 5HT-Rezeptoren. Die 5HT-Sekretion wird durch Bindung von 5HT an präsynaptische Rezeptoren beendet. Überschüssiges Serotonin wird über SERT (Serotonin-Reuptake-Transporter) rücktransportiert und anschließend entweder über die mitochondriale MAO zu 5-Hydroxyindolessigsäure (5-HIES). metabolisiert oder erneut vesikulär gespeichert. Bei Dunkelheit wird der Abbauweg hin zu Melatonin aktiviert, wobei SAMe die für die Melatoninbildung notwendige Methylgruppe liefert. Ein limitierender Faktor der Serotoninkonzentration im Gehirn ist der Tryptophangehalt in der Nahrung. Häufig wird Tryptophan-Entzug als Modell verwendet, um die klinischen Auswirkungen von Serotoninmangel und das gesamte Spektrum der physiologischen Effekte von Serotonin zu untersuchen.Die Bedeutung derf Tryptophanzufuhr zeigt sich daran, dass bereits nach 24h Trp-
Entzug deutliche Serotonin-Mangelerscheinungen auftreten (Vielhaber, 2005). Einseitige
Ernährung ist daher immer wieder Ursache von Serotonin-Mangelreaktionen. Trp-reiche
Nahrungsmittel sind vor allem Milch, Bananen, Quark, Käse, außerdem Lachs, Makrele,
Truthahn, Ente, Fasan, Avocado, Sonnenblume, Kartoffel.
Serotonin-Wirkungsspektrum
Das Wirkungsspektrum von Serotonin ist enorm breit. Interaktiv mit Dopamin und Noradrenalin
wirkt es auf Schlaf, kognitive Leistungsfähigkeit, Gedächtnisfunktion, Energiehaushalt,
Körpertemperatur, Darmmotilität, Herzkreislaufsystem, Nociception, Sexualität,
Aggressionsverhalten, Stimmungslage, Ängste (Jorgensen, 2007). Es wirkt insbesondere stark
stimmungsaufhellend, antidepressiv, angstlösend, entspannend, schlaffördernd, verbessert die
Motivation und kognitive Leistungsfähikeit und erhöht die Schmerzschwelle. Zudem beeinflusst
es direkt und indirekt über die CRH-Ausschüttung das Essverhalten, wirkt appetitregulierend,
stoppt den Heißhunger auf Kohlehydrate (Craving) und verbessert den Energiestoffwechsel u.a.
durch Höherregelung der Körpertemperatur. Serotonin-Neurone stimulieren über 5HT1A- und
5HT2-Rezptoren die hypothalamische CRH-Ausschüttung und aktivieren maßgeblich die
gesamte neuroendokrine Stressantwort (Contesse, 2000; Jorgensen, 2007). Serotonin, seine
Vorstufe 5HTP, und Serotoninagonisten aktivieren zusätzlich auf direktem Wege die
hypophysäre ACTH-Skretion. Außerdem hemmt Serotonin analog Dopamin die Sekretion von
Prolactin und Gonadotropinen (Golden 2002).
Symptome des Serotoninmangels sind Craving (Heißhunger), Gewichtszunahme, Fatigue (CFS),
Schlafstörungen, Essstörungen, Depressionen, Unruhe, Angstzustände, Panikattacken,
mangelhafte Affektkontrolle, Konzentrationsschwäche, Gedächtnisschwäche, gesteigerte
Schmerzsensitivität, Kopfschmerzen, Migräne, Fibromyalgie, Reizdarm, Empfindungsstörungen
bis zu chemischer Hypersensitivität (MCS). Gleichzeitiger Melatonin- und/oder
Katecholaminmangel (Dopamin) verstärken die Symptomatik. Serotoninmangel bedeutet
allerdings nicht zwangsläufig auch Melatoninmangel und umgekehrt, da die Synthese der beiden
Mediatoren unterschiedlich reguliert ist.
L-Glutamat
ist zusammen mit L-Aspartat das quantitativ bedeutendste exzitatorische Neurotransmittersystem
im ZNS. Glutamat (Glu) hat neben der neuronalen auch eine wichtige metabolische Funktion im
Gehirn. Glutamaterge Neurone sind vor allem im zerebralen Cortex angesiedelt, von wo aus sie
nahezu alle Hirnregionen innervieren. Es handelt sich im Regelfall um sogenannte Interneurone,
die in unmittelbarem Antagonismus zu GABA das Aktivitätsniveau praktisch aller Hirnregionen
stueern. Für Glutamat existiert ein spezifischer synaptischer Reuptake-Mechanismus (EAAT3),
über den Glu nach synaptischem Release präsynaptisch inhibitorisch oder postsynaptisch zur
erneuten Speicherung in Vesikeln aufgenommen wird. Glutamat wird hochselektiv im Austausch
gegen Cystin über einen membrangebundenen Cys-Glu-Transporter aus der Zelle entfernt.
Steigerung des Cystein/Cystinangebots führt daher zur Senkung des Glutamatspiegels in
Nervenzellen.
Glutamatrezeptoren werden nach der Bindung ihrer bekanntesten Agonisten klassifiziert. Die
ionotropen NMDA(N-Methyl-D-Aspartat)- und AMPA-Rezeptoren, die Na+-/K+- bzw. Ca++-
selektive Ionenkanäle bilden, sind am wichtigsten. Die ionenselektiven Kainat-Rezeptoren und
die metabotropen, second messenger-gekoppelten Quisqualat-Rezeptoren (mGluR1-8) sind
dagegen von geringerer Bedeutung. Die metabotropen mGlu-Rezeptoren sind vorwiegend
präsynaptisch angeordnet und modulieren die Aktivität der Glu-Neuronen. Außerdem stimulieren
sie den Glu-Uptake in Astrozyten. Die NMDA- und AMPA-Rezeptoren sind im ZNS
vorherrschend. Sie sind unter Ruhebedingungen durch Mg++ blockiert. Glycin und D-Serin wirken
aktivitätssteigernd.
Etwa 70 Prozent der exzitatorischen Impulse im ZNS gehen von Glutamat aus. Glu hat
besondere Bedeutung für motorische Funktionen (Muskelarbeit, Sinne, Koordination) und
beeinflusst die Sekretion hypophysärer Hormone (HGH, ACTH). Unverzichtbar ist Glu bei der
Verarbeitung von Sinneswahrnehmungen, bei der Ausführung von Bewegungen und für höhere
Gehirnfunktionen wie Lernen und Gedächtnis. Auch die Appetitregulation ist Glu-Einflüssen
unterworfen, es wirkt appetitsteigernd und supprimiert das Sättigungsempfinden. Es wird daher in
der Tierzucht auch als Mastmittel für schnellen Körpergewichtaufbau eingesetzt. Auch bei Depressionen ist Glutamat von Bedeutung. Glu-Antagonisten wie Lamotrigin, Ketamin, Riluzol oder Memantin, ebenso der partielle Glu-Agonist D-Cycloserin, haben antidepressive Wirkungen (Sanacora, 2003). Glycin, D-Serin und D-Cycloserin scheinen auch für die Behandlung der Schizophrenie aussichtsreich. Im Überschuss entwickelt Glutamat ausgeprägtes neurotoxisches Potential, durch Destruktion der Glutamatrezeptoren (Exzitotoxizität) und Induktion der Apoptose von Nervenzellen. Daher hat Glutamat erhebliche Bedeutung für neurodegenerative Krankheiten wie Epilepsie, Lähmungen nach Schlaganfall, Parkinson und Alzheimer. Glutamat kommt natürlicherweise in vielen wohlschmeckenden Lebensmitteln vor, u.a in Fisch, Tomaten, Käse. Ein Vielfaches der natürlich vorkommenden Menge wird allerdings als Geschmacksverstärker in der Essensherstellung verwendet. Das Chinarestaurant-Syndrom wird mit Glutamat in Verbindung gebracht, allerdings bis heute ohne Bestätigung. Der Einsatz von Glu in der Lebensmittelzubereitung gilt bisher nicht als bedenklich (DGE Prsseerklärung, 2006). Allergische Unverträglichkeitsreaktionen gegenüber Glutamat sind allerdings beschrieben. Glutamat kann endogen aus Ketoglutarat im Citratzyklus unter Mitwirkung der GAD (Glutamat-Dehydrogenase) und Ammoniak hergestellt werden. Es dient einerseits als Vorstufe seines wichtigsten Gegenspielers im ZNS, GABA, andererseits wird unter Mitwirkung der Glutaminsynthase (GlnS) Glutamin gebildet. Die GAD- und GlnS-Reaktion sind beide von besonderer Bedeutung als Entgiftungsreaktionen für das ZNS, wobei Ammoniak „entgiftet" wird.
Glutamat entsteht im Citratzyklus aus α-Ketoglutarat (αKG) und einem Ammoniumion durch die Reaktion der Glutamat-Dehydrogenase (GDH). Ein weiteres Ammoniumion kann über die Reaktion der Glutamin-Synthase (GlnS) abgefangen werden, wobei Glutamin entsteht. Beide Reaktionen dienen der spontanen Entgiftung aller Gewebe und sind im Gehirn von besonderer Bedeutung. Für die endgültige Entgiftung müssen Ammoniumionen dem Harnstoffzyklus zugeführt werden. Dies erfolgt sowohl durch Übertragung auf Oxalacetat (OA), als auch über die Glutamat-Dehydrogenase Reaktion. Glutamin kann mit α-Ketoglutarat zu zwei Molekülen Glutaminsäure umgesetzt und damit der GDH-Reaktion zugeführt werden. Diese Reaktion wird durch Glutamat-Synthase (GluS) katalysiert. Bei der Aminosäuresynthese ist Glutaminsäure der NH2 Donor in einer Transaminierungsreaktion. Diese überführt α-Ketosäuren in die homologen α-Aminosäuren. Beispiele sind Glutamat-Oxalacetat-Transaminase (GOT) und Glutamat-Pyruvat-Transaminase (GPT). Coenzym ist Pyridoxal-phosphat. Bei der Für nahezu alle anderen Aminogruppen, die im Stoffwechsel benötigt werden, ist Glutamin der Donor.
GABA
Gamma-Aminobuttersäure (GABA) ist der wichtigste inhibitorische Neurotransmitter des
Zentralnervensystems. Nach Glutamat, dem wichtigsten exzitatorischen Neurotransmitter, ist die
GABA-Konzentration im ZNS am höchsten. Paradoxerweise werden beide, GABA und Glutamat,
aus derselben Aminosäurevorstufe gebildet. GABA wird vorwiegend über den sog. GABA-Shunt
synthetisiert. Ketoglutarat aus dem Krebszyklus wird zunächst enzymatisch (GLDH) mit
Ammoniak zu Glutamat umgewandelt, das dann in GABA-Neuronen durch GAD (Glutamat-
Decarboxylase), Kofaktor PLP (Pyridoxal-5-Phosphat/Vitamin B6), zu GABA umgewandelt wird. GABA kann über die GABA-Transaminase wieder zu Glutamat aufgebaut und weiter zu Succinat-Semialdehyd metabolisiert werden. Oder Glutamin dient als GABA-Ausgangspunkt, das über Glutamat (Glutaminase) zu GABA decarboxyliert wird (GAD). Glutamat übernimmt in diesen Reaktionen die Entgiftung von Ammoniak, das in den Harnstoffzyklus übertragen wird. Erst um 1970 wurde die herausragende Bedeutung von GABA als inhibitorischer Neurotransmitter erkannt. Es wirkt bei zahllosen neuronalen Vorgängen im ZNS modulierend mit, meistens unmittelbar antagonistisch gegenüber dem exzitatorischen Glutamat. 30 - 40% aller neuronalen Synapsen sind GABAerg. Die meisten dieser GABA-Neurone sind sog. Interneurone, die die Aktivität anderer, vorwiegend mit biogenen Aminen operierender Neurone kontrollieren. Daneben existieren jedoch auch effektorische, in die Peripherie projizierende GABAerge Neurone. Auch primär periphere GABA-Neurone sind heute bekannt, vor allem im enteralen Nervensystem. Die Hauptkonzentration GABAerger Interneurone findet sich im Thalamus, Hippocampus und zerebralen Cortex. GABAerge Zentren sind außerdem im Striatum (95% GABAerge Neurone), im Globus pallidus, Cerebellum und in der Substantia nigra vorhanden. Drei Klassen von GABA-Rezeptoren sind bekannt, GABAA-C. Benzodiazepine und Barbiturate wirken akzessorisch über
den GABAA-Rezeptor und verstärken die GABA-Wirkung. Bedeutende GABA-Enhancer sind die
intracerebral synthetisierten oder über die BHS importierten Pregnan-Steroide. Der Schlaf-fördernde und sedierende Effekt von oralem Progesteron beruht maßgeblich auf seiner GABAA-
Rezeptoraffinität. Noch stärker wirksam ist sein bei oraler Gabe in der Leber bzw. im ZNS selbst gebildeter Hauptmetabolit allo-Pregnanolon. Besondere Bedeutung für die Wirksamkeit von GABA hat auch Serotonin, das die GABA-Synthese stimuliert und die GABA-Rezeptoraffinität erhöht (Jorgensen, 2007). Bei Serotoninmangel ist auch die Wirksamkeit von GABA eingeschränkt. Weitere GABA-Mimetika sind Theanin, Taurin und Rhodiola, die ebenfalls am GABA-Rezeptor angreifen und die GABA-Wirkung verstärken. Besondere Bedeutung für die Wirkung von GABA hat Serotonin, das die GABA-Synthese stimuliert und die GABA-Rezeptoraffinität verbessert. Bei Serotoninmangel ist auch dieWirksamkeit von GABA eingeschränkt. Weitere GABA-Mimetika sind Theanin, Taurin und Rhodiola, die ebenfalls am GABA-Rezeptor ansetzen und die GABA-Wirkung verstärken. Taurin wirkt allerdings stärker über Glutamat-Hemmung. GABA wirkt in erster Linie durch Hemmung der präsynaptischen Freisetzung exzitatorischer Neurotransmitter, meist in unmittelbarem räumlichem Antagonismus zu Glutamat. Es hemmt die CRH-ACTH-Cortisol-Stressachse und die hypophysäre Gonadotropinsekretion, während Glutamat aktivierend wirkt (Hermann, 2004). Die Aktivierung exzitatorischer Neurone wird durch Steigerung der GABA-Synthese gegenreguliert. GABA wirkt anxiolytisch, analgetisch, relaxierend, antikonvulsiv und blutdruckstabilisierend. Außerdem besitzt GABA eine noch über Serotonin und Melatonin hinausreichende schlaffördernde Wirkung. Sehr niedrige GABA-Konzentrationen werden bei gravierenden Störungen des Neurotransmitter-Netzwerks, Hochdruck, chronischen Schmerzen, irritablem Kolon, prämenstruellem Syndrom und Depressionen gefunden. Bei Epilepsie, Schizophrenie, Manie und schweren Depressionen ist GABA im Blut konstant niedrig und bleibt auch unter Therapie unverändert niedrig, sodass es als biologischer Marker diskutiert wird (Petty, 1994). Komplikationen des GABA-Mangels sind Heißhunger auf Zucker/Süßigkeiten, Parästhesien, Muskelverspannungen, Ohrgeräusche (Tinnitus), veränderte Geruchsempfindungen, nächtliches Schwitzen, Hyperventilation, Tachykardien, Gedächtniseinbußen, Impulsivität, Ungeduld, Ängste. Vor allem die angstlösende Wirkung von GABA wird klinisch genutzt. Da jedoch GABA selbst die Blut-Hirnschranke nicht passieren kann, wurden lipophile GABA-Derivate wie Gabapentin oder Pregabalin entwickelt, die als First-line Medikation für das generalisierte Angstsyndrom gelten. Neben seinen neuronalen Wirkungen hat GABA vielfältige parakrine und endokrine Wirkungen. Es wirkt zentral auf die hypothalamische Sekretion von Releasing-Faktoren, GABAerge Neurone innervieren die Hypophyse, GABA wird parakrin über das Pfortadersystem zur Hypophyse
transportiert und es wird in der Hypophyse selbst synthetisiert, wo es die Produktion von Prolactin, ACTH, TSH und LH hemmend moduliert. Vor allem aber stimuliert GABA die Wachstumshormonsekretion über Aktivierung des hypothalamischen HGH-Releasing-Hormons (GHRH) und unmittelbar lokal in der Hypophyse. Auch in den Pankreas-Inselzellen wird GABA lokal produziert und moduliert die Insulinsekretion. Für die Behandlung des GABA-Mangels empfehlen sich mehrere Möglichkeiten:
1. Substitution mit der Glutamat/GABA-Vorstufe Glutamin, das zudem für die Entgiftung des
ZNS eminent wichtig ist. W
2. Behandlung mit Glutamin in Kombination mit Glycin, der kleinsten Aminosäure, die
überwiegend GABA-artig wirkt. Glutamin/GABA und Glycin wirken synergistisch schlaffördernd, entspannend und bahnend für die nächtliche Regeneration des Endokriniums.
3. GABA selbst hat bei oraler Gabe zwar infolge Blockade durch die Blut-Hirnschranke nur
marginale zentrale Effekte, seine peripheren Wirkungen auf endokrine Organe und Immunsystem sind jedoch nicht beeinträchtigt – soweit sie durch parakrines GABA hervorgerufen werden.
4. Begrenzte zentrale Effekte durch GABA selbst können mit modifizierten sublingualen
GABA-Präparaten erreicht werden, die über die Mundschleimhaut „auf Nebenwegen" ins Gehirn gelangen können. Auf Grund ihrer schnellen, innerhalb von Minuten einsetzenden Wirkung, sind sie für die Sofortbehandlung von Angstzuständen geeignet.
5. Schließlich existieren lipophile GABA-Derivate, die starke GABA-Wirkung aufweisen. Z.b.
GABA gekoppelt an Niacin (
Kavinace ) oder GABA gekoppelt an Phenol (Phenibut ).
6. Zahlreiche Pharmaka leiten sich strukturell von GABA ab und sind im Vergleich zu GABA
besser BHS-gängig. Die lange bekannte Valproinsäure (Ergenyl®) und Vigabatrin (
Sabril ) verstärken die GABA-Wirkung durch Hemmung des Abbaus (Hemmung der
GABA-Transaminase), Steigerung der GABA-Synthese und durch Reuptakeblockade (Vigabatrin). Progabid ist eine ZNS-gängige GABA-Variante, aus der im Gehirn GABA freisgesetzt wird (nur in Frankreich als
Gabrene erhältlich). Die aktuellsten
Entwicklungen sind Pregabalin (
Lyrica ) und Gabapentin (Neurontin ), deren Wirkung
jedoch nicht auf einen direkten GABA-Mechanismus, vielmehr auf Glutamat-antagonistische Effekte zurückgeführt wird.
NEUROMODULATOREN
Taurin
Taurin ist im engeren Sinne keine Aminosäure sondern eine Aminosulfonsäure, die aus Cystein
(Methionin) unter Mitwirkung von Vitamin B6 gebildet wird. Über die Nahrung wird Taurin
ebenfalls in höheren Mengen zugeführt, sodass nur bei hohem Verbrauch (Leistungssport,
Leberkrankheiten, Arteriosklerose, Augenkrankheiten, oxidativer Stress) Mehrbedarf entstehen
kann. Die größten Taurinkonzentrationen finden sich im ZNS, der Retina, den Thrombozyten,
Granulozyten und der Muskulatur.
Taurin ist peripher bei der Gallensäurekonjugation beteiligt, wirkt als starkes Antioxidans ohne
prooxidatives Potential, ist in die Entgiftung eingebunden und wirkt wachstumsfördernd. Taurin ist
kein Neurotransmitter, wirkt jedoch über die Stimulation des Calciuminflux und
Membranstabilisierung antiarrhythmisch, antiepileptisch und zusammen mit Glutamat ZNS-
entgiftend. Peripher (Gallensäurekonjugation) und im ZNS wirkt Taurin synergistisch mit GABA
und Glycin. Zusammen mit GABA und Glycin unterstützt Taurin inhibitorische Signale und wirkt
beruhigend, angst- und krampflösend. Außerdem durch Steigerung des Acetylcholin-Gehalts im
Gehirn kognitiv leistungssteigernd. Taurin steht in Wechselbeziehung mit Melatonin. Während die
Melatoninsynthese bei Dunkelheit ansteigt, wird Taurin in der Epiphyse bei Tageslicht gebildet.
L–Theanin
Theanin ist eine Glutamin-ähnliche Aminosäure, die fast ausschließlich in der Teepflanze
vorkommt (1 - 2% Gewichtsanteil). L-Theanin wird gut über den Dünndarm resorbiert und gelangt
über den Carrier für neutrale Aminosäuren durch die Blut-Hirn-Schranke, wo es etwa 5 Stunden
nach Aufnahme seine maximale Konzentration erreicht. Bei der Ausscheidung über die Nieren
wird Theanin zu Glutamat (Glutaminsäure) hydrolysiert.
Im Gehirn steigert Theanin die Dopamin-Produktion, senkt Noradrenalin und blockiert die
Glutamataktivität durch Hemmung der Rezeptorbindung von Glutamat und Reuptakeblockade im
Hippocampus. Die Synthese und die Wirkung von GABA werden verstärkt. Serotonin wird in
einigen Hirnregionen (Hippocampus, Hypothalamus, Striatum) stimuliert, in anderen Regionen
jedoch gesenkt. Durch seine ausgeprägte antioxidative Wirkung schützt Theanin gesunde Zellen
vor oxidativen Schäden und erhöht intrazelluläres Glutathion.Theanin wirkt mäßig beruhigend,
entspannend, stresslösend und anxiolytisch.
Vitamin C
Ascorbinsäure (Vitamin C) wird ebenfalls zu den Neuromodulatoren gerechnet (Castro, 2009). Es
moduliert die Aktivität dopaminerger und glutamaterger Neurone, wirkt als Kofaktor bei der
Synthese von Noradrenalin und verschiedenen Neuropeptiden und bei der synaptischen
Freisetzung von Noradrenalin und Acetylcholin. In höherer Konzentration (> 1 g tgl) hat es
darüber hinaus antidepressives Potential (Binfare, 2009). Ascorbinsäure wird über das Liquor-
Blut-Interface durch einen spezifischen Na-Vitamin C-Transporter (SVCT), alternativ auch über
die BHS als Dehydroascorbinsäure durch Glukosetransporter GLUT1-4 im Gehirn bis zu
millimolaren Konzentrationen angereichert (Agus, 1997). Seine Konzentration wird mit hohem
Aufwand konserviert. In Stressituationen reduziert Ascorbat den Cortisolanstieg und führt zu
rascherem Abfall in der Erholungsphase (Brody, 2002). Es ist Kofaktor bei zahlreichen
Reaktionen, neben der Synthese von Katecholaminen auch für Carnitin und für Myelin.
Ascorbinsäure, die im Gehirn als herausragend wichtiges Antioxidans fungiert und die Integrität
von Neuronen sichert, wird lokal in erster Linie über Glutathion in Astrozyten recycelt. Neue
Untersuchungen sprechen für Ascorbinsäure als metabolischen Schalter, der die neuronale
Energiegewinnung bei Aktivitätsanstieg von Glukose, dem Standardsubstrat des Gehirns, zu
Lactat umschaltet. Lactat wird von Glutamat-aktivierten Astrozyten bereitgestellt denen
offensichtlich zentrale Bedeutung für die Regulation der metabolischen Aktivität im Gehirn
zukommt (Castro, 2009).
Vitamin D3
Neben seiner traditionellen Rolle in der Calciumhomöostase und im Knochenstoffwechsel hat
Vitamin D3 (VD3) zahlreiche weitere Funktionen, die es bis heute zu einem der wichtigsten
Hormone überhaupt werden ließen. Es wirkt auf zellulärer Ebene wachstumsregulierend,
verbessert den Kohlehydratstoffwechsel, wirkt antientzündlich, immunregulierend, hemmt
überschießende Th1-Immunreaktivität und Autoimmunität, steigert die Infektresistenz. Fast alle
Körperzellen verfügen über Vitamin D-Rezeptoren und sind in der Lage, aus der inaktiven
Vitamin D3-Vorstufe Cholecalciferol (25[OH]-Vitamin D3) durch lokale alpha-Hydroxylasen das
aktive Calcitriol (1,25[OH]2-Vitamin D3) zu synthetisieren. Neuerdings wird VD3 auch zu den
Neurosteroiden gerechnet. Im Gehirn finden sich sowohl Rezeptoren für VD3 als auch für
Parathormon, außerdem Hydroxylasen für die Aktivierung von VD3 zu Calcitriol.
Bei der großen Mehrheit der westlichen Bevölkerung besteht ein erheblicher Vitamin D-Mangel,
da die körpereigene Synthese mangels UV-Lichteiwirkung auf die Haut ungenügend ist und die
alimentäre Aufnahme aus Meeresfischen (Vitamin D3 kommt fast nur in maritimen Produkten vor)
zu gering ist. Heute wird als untere Normgrenze eine Blutkonzentration von 30 ng/ml Vitamin D3
(Cholecalciferol) angesehen. Bei Unterschreiten dieser Grenze kommt es zum
kompensatorischen Anstieg von Parathormon, der bes zum sekundären Hyperparathyroidismus
gehen kann (Jorde, 2006).
Niedrige VD3-Spiegel sind mit depressiver Stimmungslage assoziiert, VD3-Substitution
verbessert die Stimmungslage. Zahlreiche Untersuchungen haben außerdem gezeigt, dass
Vitamin D-Mangel mit kognitiven Einbußen, Neurodegenration und erhöhter Demenzrate
korreliert.
STRESS-IMMUNACHSE
Das Immunsystem bildet neben der Stresshormonachse und dem zentralen und peripheren
autonomen Nervensystem als IRS (Immun-Response-System) die vierte Achse der
Stressbewältigung. Bestandteil jeder Stressreaktion ist eine zeitlich begrenzte Entzündungsreaktion mit Ausschüttung von proinflammatorischen Zytokinen wie TNF-alpha, Interleukin-1ß, Interleukin-6 oder Interferon-gamma, während adaptive zelluläre Immunfunktionen (T- und NK-Zellaktivität) blockiert werden. Hauptquelle der zentralen Entzündungsmediatoren sind die Mikrogliazellen, die ihrerseits immunologischen Ursprungs sind (Makrophagen) und ca 10% des polyvalenten Neuroglia-Netzwerks im ZNS darstellen (Barres, 2008). Gliazellen stellen die Mehrheit der Zellen des zentralen Nervensystems, sie machen insgesamt 70% der Hirnzellen aus (Farooqui, 2007).Auch die anderen Gliazellen (Astrozyten/Astroglia; Oligodendrozyten) und die Nervenzellen selbst sind in der Lage, proentzündliche Zytokine und Chemokine zu synthetisieren und in Stresssituationen zu sezernieren. Im entzündlichen Milieu werden die langkettigen, polyungesättigten Omega 3(w3)-Fettsäuren DHA, EPA und die w6-Arachidonsäure (AA) aus den Phospholipiden der Zellmembranen durch Phospholipasen (PLA2), die ihrerseits über TNFα oder ROS aktiviert werden, herausgelöst
(Calder, 2006). Über COX2 und 5-LOX werden aus der Arachidonsäure proentzündliche Prostanoide und Prostaglandine (PgE ) bzw. Thromboxan A2, Leukotriene
als Eicosanoide bezeichnet werden, generiert. Die AA-Metaboliten besitzen proentzündliche,
immunsuppressive und gerinnungsfördernde Aktivität, wirken bronchokonstriktorisch,
ödemfördernd und neurotoxisch (Farooqui, 2007; Serhan, 2007).
Im Unterschied zur AA werden aus den w3-Fettsäuren antientzündlich, antiproliferativ und
antithrombotisch wirksame Metaboliten gebildet. Sie sind wichtig für die Downregulation von
Entzündungen, bremsen oxidativen Stress, wirken antiapoptotisch durch Stimulation der Bcl-
Apoptoseinhibitoren und Hemmung der proapoptotischen Mediatoren BAX und BAD.
Je höher der Anteil der w3-Fettsäuren DHA und EPA gegenüber der w6-FFS AA in den
neuronalen Membranlipiden ist, desto weniger proentzündliche Metaboliten werden bei
entzündlichem oder oxidativem Membran-Breakdown freigesetzt.
Die Entzündungsreaktion auf Stress kann höchst unterschiedliche Qualität besitzen. Während sie
normalerweise nach Dauer und Ausmaß begrenzt ist, reagieren einige Individuen erheblich
länger und stärker entzündlich auf akuten Stress (Bierhaus 2003).
Abbildung: Entzündliche Zytokinreaktion bei ansonsten gesunden Probanden im Trierer Stresstest mit
Prüfungssimulation:
Diese Personen entwickeln bei anhaltendem Stress das sog. „Sickness-Behavior", die
pathologische Form der Stressreaktion, die durch Inappetenz, Temperaturanstieg, Fatigue,
Schmerzen, Akut-Phase-Reaktionen, Stimmungsschwankungen bis zu Ängsten und
Depressionen und durch Schlafstörungen geprägt ist (Dantzer, 2009). Während bei der
physiologischen Stressreaktion die neuroendokrinen Anpassungsmechanismen dominieren
(Stresshormone, Katecholamine, Serotonin) sind die proinflammatorischen Zytokine, vor allem IL-
1ß und IL-6, für die pathologische „Sickness"-Adaptation bestimmend und maßgeblich an der
Entwicklung der zentralen „Fatigue" beteiligt.
Zytokinwirkungen im ZNS
Die produktive Rolle, die den proinflammatorischen Zytokinen in der Stressantwort zukommt,
wurde in den letzten Jahren zunehmend deutlicher. Seit längerem ist bekannt, dass Zytokine in
der akuten Stresssituation synergistisch mit anderen Akteuren des Stressbewältigungssystems
die neuroendokrine Stressantwort stimulieren. IL-1ß und IFN-gamma steigern die Ausschüttung
von CRH und ACTH und potenzieren die Stresshormonsekretion. Die Wirkung von IL-1ß auf die
Stressachse übertrifft selbst die von CRH. Die Zytokine der Entzündungskaskade wirken
außerdem auf die Neurotransmission und potenzieren Sekretion und Turnover von Dopamin,
Noradrenalin, Serotonin, GABA und Acetylcholin (Tabelle).
Über ihre neuroendokrin stimulierenden Effekte hinaus sind Zytokine zentral an vielen
fundamentalen Stressadaptationsmechanismen maßgeblich beteiligt. Generell trifft für die
Zytokineffekte zu, dass sie stark dosisabhängig sind. Bei kurzzeitigem stressbedingtem
Konzentrationsanstieg stehen positive stimulierende und trophische Effekte im Vordergrund
während längere Zytokineinwirkung sich meist negativ sowohl auf mentaler, psychischer,
vegetativer als auch auf struktureller Ebene auswirkt und Neurodegeneration fördert. Die
Stressentzündung stimuliert intermittierend die Sekretion neuronaler Wachstumsfaktoren wie
BDNF (Brain-derived Neurotrophic Factor), NGF (Nerve Growth Factor) oder IGF-I, die
Ausreifung neuronaler Progenitorzellen, die dentritische Vernetzung von Neuronen sowie die
Replikation und Differenzierung von Gliazellen. Die starke Zytokininduktion bei Infekten im
Kindesalter, die am alterstypisch hohen Fieberanstieg, Muskelschmerzen und exzessiver
Müdigkeit bei Infekten ablesbar ist, hat daher eine überaus wertvolle Triggerfunktion für die
Reifung des ZNS. Proinflammatorische Zytokine können außerdem kurzfristig die kognitive
Leistungsfähigkeit, Wachheit, Konzentration, Lern- und Gedächtnisleistung erhöhen. Dadurch
wird u.a. auch die Reaktionsbereitschaft gegenüber Gefahren optimiert, ebenso die
Gedächtnisformierung nach Gefahrsituationen, die umso dauerhafter im Gedächtnis verankert
wird je extremer die zu erinnernde Situation.
Dauernder Stress wirkt dagegen über inflammatorische und neuroendokrine Faktoren (Cortisol,
Glutamat, oxidativer Stress, Fettsäuremetaboliten) hemmend auf das Kurzzeitgedächtnis,
beeinträchtigt das Konzentrationsvermögen und die Lernfähigkeit. Darüber hinaus können
psychische Veränderungen ausgelöst oder verstärkt werden, die sich u.U. erst mit
fortschreitendem Alter manifestieren.
NEUROENDOKRINE GESUNDHEITSSTÖRUNGEN
Eine enorme Vielfalt gesundheitlicher Probleme ist direkt oder indirekt mit gravierenden
Störungen neuroendokriner Funktionsabläufe, gestörter Neurotransmittersynthese und Störungen
der Neurotransmitter-Balance verbunden. Nicht selten auch mit gestörter Immunfunktion oder
gesteigerter entzündlicher Aktivität. Zu den Problemen zählen scheinbar so unterschiedliche
Syndrome wie:
Adipositas/Übergewicht, Essstörungen (Essattacken, Heißhunger), Schlafstörungen, Insomnie, depressive
Verstimmung bis zu reaktiven Depressionen, Majordepression, bipolare, manische Depression, saisonale
Gemütsschwankungen (SAD: saisonal affective Disorder), Schizophrenie, Epilepsien, Angststörungen,
Panikattacken, Erschöpfung/Fatigue (CFS: chronisches Fatigue-Syndrom), Aufmerksamkeitsstörungen
(ADS/ADHS), Fibromyalgie, MCS/Multiple chemische Sensitivität, Migräne, irritables Colon/Reizdarm,
Stress, Burn-Out Syndrom, posttraumatisches Stresssyndrom (PTSD), prämenstruelles Syndrom (PMS),
Menopause-Syndrom, Parkinson-Syndrom.
Chronischer Stress
Die endokrine Stressachse (Hypothalamus-Hypophyse-Nebennierenrinde) ist bei chronischem
Stress dauerhaft aktiviert. CRH, ACTH und Cortisol sind erhöht. Der Cortisol-Tagesrhythmus ist
intakt, die Cortisol-Gesamtproduktion jedoch insgesamt erhöht und der morgendliche
Cortisolspiegel deutlich höher als normal. Im Unterschied zum Cushing-Syndrom bleibt jedoch
die Morgen-Abend-Differenz der Cortisolwerte erhalten. Im Zweifelsfall kann durch den
Dexamethason-Hemmtest eine Abgrenzung zum echten Cushingsyndrom vorgenommen
werden. Bei fortdauernder Aktivierung der HPA-Achse wird der physiologische Cortisol-
Tagesrhythmus zunehmend gestört. Die nächtliche, regenerative Zunahme der
Cortisolproduktion kann zurückgehen, der morgenliche Cortisolwert absinken und im
Tagesverlauf können starke Konzentrationsschwankungen auftreten. Bei genetisch suszeptiblen
Personen kann die Affinität der Glukokortikoidrezeptoren (GR) abnehmen und die
Cortisolwirkung bei andauerndem Hypercortisolismus nachlassen. Durch den chronischen
Cortisolüberschuss wird die Serotoninsynthese gehemmt, die Aktivität des cholinergen Systems
reduziert und peripher überwiegt der Sympathikotonus. Der Cortisolexzess wirkt außerdem
neurotoxisch durch verstärkte Apoptose von Nervenzellen und Hemmung der Neuroregeneration
mit Abfall des Neurogenese-Faktors BDNF. Die Immunfunktion wird gravierend verändert:
Hemmung der zellulären Immunität mit Th2-Shift und Verstärkung der humoralen Immunantwort,
Entzündungshemmung, deren Effizienz allerdings mit Dauer der Stresskonstellation abnimmt.
Burn-Out
Das Burn-Out-Syndrom stellt eine tiefgreifende Störung der Produktion von Stresshormonen
(Cortisol, Adrenalin) und Neurotransmittern (Serotonin, Noradrenalin) als Folge langanhaltender
Belastung dar, die individuell zur Überforderung und zum Zusammenbruch der
Kompensationsmechanismen führt. Voraussetzung für diesen fatalen Verlauf der Stressreaktion
sind offensichtlich genetisch disponierende Individualfaktoren, die u.a. die Syntheseleistung, die
Metabolisierungsrate und die Rezeptoreigenschaften der neuroendokrinen Signalsysteme
betreffen. Zwei Schwerpunkte prägen die Pathophysiologie des Burn-Out Syndroms: Der
Zusammenbruch der physiologischen Balance des Stresshormon- und Neurotransmitter-
Haushaltes und die gesteigerte inflammatorische Aktivität. In mehreren Untersuchungen wurde
die basale und stimulierte Cortisosekretion im Speichel bei Burnout-Patienten gemessen (Grossi,
2005; Osterberg, 2009).
In fortgeschrittenen Fällen geht der 24h-Rhythmus der Stresshormonproduktion verloren, die
nächtliche Aktivitätszunahme der HPT-HVL-NNR-Achse flacht ab bis sie völlig abklingt und der
morgendliche Cortisolwert sinkt auf ein Minimum ab. Im Tagesverlauf bleibt die HVL-NNR-
Aktivität niedrig, der Cortisolwert steigt kaum über das morgendliche Minimum hinaus. Häufiger
sind allerdings die offensichtlich weniger ausgeprägten Fälle mit „chaotischer" Cortisol-
Tageskurve und Anstieg im Sinne einer „Erholung" im Ablauf der Tagesbelastungen. Auch die
Hypophysenaktivität ist bei fortgeschrittenem Burnout niedrig. D.h. auch die Hypopyhse ist
betroffen, da andernfalls bei Absinken der NNR-Aktivität ACTH wegen der Feedback-Kopplung
der Stresshormonachse ansteigen müsste. Wahrscheinliche Ursache ist eine Desensibilisierung
der HPT-HVL-Achse mit Blockade oder Verlust der hypophysären CRH1-Rezeptoren. Alternativ
kommt auch Aktivitätsabfall des Hypothalamus in Betracht, allerdings ist nahezu nichts über die
zentrale CRH-Aktivität beri Burnoutv bekannt. Auch Melatonin, einer der zentralen Taktgeber der
Hormonzyklen, der ebenfalls in der Nacht gebildet wird, ist nicht mehr ausreichend verfügbar.
Dies ist u.a. auf den Serotoninmangel zurückzuführen, der sich als Folge gesteigerten
Verbrauchs und der Synthesehemmung entwickelt. Auch Noradrenalin und Adrenalin sind meist
erniedrigt.
Diagnostisch weisen sehr niedrige morgendliche Cortisolwerte im Speichel und ggf. eine
gestörte Tagesrhythmik auf das Burn-Out-Syndrom hin. Die Feststellung eines gleichzeitig
niedrigen ACTH belegt, dass es sich um eine zentrale, der Hypophyse vorgelagerte Blockade der
HVL-NNR-Achse handelt und nicht um eine organische Insuffizienz der Nebennierenrinde. Das
Tagesaktivitätsmuster der Burn-Out Betroffenen korreliert mit dem Tagesverlauf des Cortisols.
Nach niedrigen Morgenwerten kann es im Lauf des Tages zu Aktivitätssteigerung mit parallelem
Anstieg des Cortisols kommen.
Der NA/A-Quotient ist infolge erhöhtem Noradrenalin hoch - bei fortgeschrittener
sympathoadrenaler „Erschöpfung" jedoch zunehmend auf sehr niedrigem Konzentrationsniveau.
Dopamin ist meist unverändert. Serotonin ist in der Regel ebenfalls mehr oder weniger stark
erniedrigt, wofür Synthesehemmung, erhöhte IDO-Aktivität und gesteigerter Substratverbrauch
verantwortlich sind.
F a t i g u e
Eines der wichtigsten und am meisten verbreiteten Leitsymptome der ins Pathologische
übergehenden Stressreaktion ist die zentrale Erschöpfung (central Fatigue), die von der
peripheren Form (peripheral Fatigue) mit muskulärer Erschöpfbarkeit zu unterscheiden ist.
Erschöpfbarkeit ist am ehesten zu definieren als die erheblich eingeschränkte Fähigkeit,
willentliche Aktionen aufzunehmen oder über längerer Zeit durchzuhalten. Zentrale
Erschöpfbarkeit betrifft sowohl die körperliche als auch die mentale und emotionale
Leistungsfähigkeit. Die Erschöpfungsreaktion entsteht auf komplexer Grundlage. Sie ist
einerseits mit den Zytokin-basierten Entzündungsreaktionen assoziiert, da proentzündliche
Zytokine wie IL-1ß, TNF-alpha oder IL-6 unmittelbar entsprechende neurologische
Komplikationen auslösen können. Andererseits mit der unter andauernder Belastung
entstehenden neuroendokrinen Dysbalance. Die entzündliche Aktivität kann außer durch
pathologischen Stress auch durch Infektionen, Autoimmunerkrankungen oder toxische Einflüsse
(Medikamente, Fremdstoffe, Strahlung) zustande kommen.
Zentrale Erschöpfbarkeit findet sich symptomatisch bei verschiedensten neurologischen
Erkrankungen, u.a. bei MS, myotoner Dystrophie, M. Parkinson, Guillan-Barré-Syndrom, nach
Enzephalitiden (Neuroborreliose, Q-Fieber, Poliomyelitis), bei zerebralen Vaskulitiden oder
Motoneuron-Erkrankungen; als postvirale Müdigkeit (Herpesvirusgruppe), bei raktivierten
Infketionen, bei Lupus, rheumatoider Arthritis oder bei Tumorerkrankungen unter/nach
Behandlung sowei bei theraputischer anwendung von Zytokinen (MS, Hepatitis, Tumoren).
C F S
Die idiopathische Form der Fatigue ist das chronische Müdigkeits- bzw. Erschöpfungssyndrom
(CFS). Das beim CFS nahezu durchgehend vorhandene Kriterium ist die Störung der
neuroendokrinen Funktionsachse mit Hypocortisolismus, der bei Frauen häufiger als bei
erkrankten Männern vorkommt (Nater, 2007). Der morgendliche Cortisolspiegel (Speichelcortisol)
ist erniedrigt, die Tagesrhythmik der Cortisolsekretion ist mehr oder weniger stark gestört. ACTH
ist ebenfalls niedrig, sodass von einer Störung auf hypothalamischer Ebene auszugehen ist.
Die hypothalamische CRH-Sekretion ist infolge anhaltender psychischer, physischer oder
toxischer Belastung dauerhaft gesteigert, die CRH-abhängigen Organe, Hypophyse und
Nebennierenrinde sind möglicherweise durch Rezeptor-Downregulation und Desensibilisierung
refraktär geworden. Individuell disponierende, genetische, Faktoren sind mit ausschlaggebend für
die Entwicklung der zentralen Fatigue. Beim CFS wurde in Zwillingsuntersuchungen eine
Konkordanz von > 50% gefunden. Auch die Neurotransmitterbalance ist gestört, vor allem
Serotonin und Noradrenalin sind erniedrigt. Bei einem Teil der CFS-Fälle spielen offensichtlich
auch protrahierte, reaktivierte Infektionen, toxisch-inflammatorische Zustände oder
schwerwiegende oxidative/nitrosative Schäden mit Hemmung der Mitochondrienfunktion nd
zentraler Enzymsysteme eine dominante Rolle
Auch das M C S (Multiple Chemische Sensitivität) ist durch einen allerdings in der Regel latenten
Hypocortisolismus gekennzeichnet. Die Ruhekonzentrationen von Cortisol und ACTH sind meist
unauffällig, unter Belastung kommt es jedoch statt zum Anstieg zum paradoxen Abfall der
Stresshormone, sodass auch hier von einer erheblichen Störung der HPT-HVL-NNR-Achse
ausgegangen werden muss. Häufig ist auch beim MCS Serotonin vermindert und der
Noradrenalin:Adrenalin-Quotient zu Noradrenalin hin verschoben.
F i b r o m y a l g i e
CFS und MCS werden heute als Varianten einer umfassenderen Gesundheitsstörung, des C M I
(Chronic Multisystem Illnesses) gesehen, zu dem auch die Fibromyalgie (FMS) gerechnet wird.
Über 40 % der FMS-Patienten sind auch von Fatigue betroffen, viele CFS-Patienten leiden auch
unter erhöhter Unverträglichkeit gegenüber Umweltstoffen (MCS) oder anderen
Umwelteinflüssen (ESM: Elektrosmog). Allen drei Manifestationsformen sind starke
Schlafstörungen gemeinsam. Bei FMS findet sich wie beim CFS und bei MCS häufig eine
gestörte Cortisol-Chronizität und inadäquate HPT-NNR-Aktivierung unter Belastung. Bei der
Mehrheit der Patienten besteht ein ausgeprägter Serotoninmangel.
Die Schmerzsymptomatik ist meist nicht gesteigerter Aktivität proentzündlicher Zytokine
zuzuschreiben, sondern weitgehend auf neuroinflammatorische Mediatoren wie Substanz P,
Neurokinine und die gestörte Neurotransmitterbalance zurückzuführen. Besonders verbreitet und
ausgeprägt sind der Serotonin- und Melatoninmangel., die meist auch Konsequenzen für andere
Exponenten der HPT-HVL-Achse wie Prolactin und Wachstumshormon haben.
Das p o s t t r a u m a t i s c h e S t r e s s y n d r o m (PTSD: „Post traumatic Stress
Disorder") ist ebenfalls durch gravierende Störungen der HPT-HVL-NNR-Achse gekennzeichnet,
die sich wie bei Fatiguesyndromen oder Burnout als Unterfunktion der CRH-ACTH-Cortisol-
Achse und der exzitatorischen Neurotransmission manifestieren.
D e p r e s s i o n
Gesteigerte HPT-HVL-NNR-Aktivität ist das häufigste neuroendokrine Kriterium der primären,
endogenen Depression, der Majordepression. Der morgendliche Cortisolwert ist erhöht und auch
im weiteren Tagesverlauf die Cortisolausschüttung hoch, u.U. zu verwechseln mit dem echten
Cushingsyndrom (psychogenes oder Pseudo-Cushing Syndrom). Der Hypercortisolismus der
Majordepression basiert auf erhöhter Aktivität der hypothalamischen Zentren, daher sind auch
ACTH und CRH erhöht. Neben genetischen Faktoren werden frühkindliche Stresstraumata mit
Veränderung der neuronalen Schaltung als strukturelle Voraussetzungen angenommen. Der
Hypercortisolismus ist einer der treibenden Faktoren der Depression (s.a. Cushing-Syndrom,
Cortisontherapie).
Außer der HPT-HVL-NNR-Achse sind bei der primären Depression häufig auch exzitatorische
Neurotransmittersysteme (Katecholamine) aktiviert. Die Affinität monoaminerger Rezeptoren ist
jedoch reduziert, sodass trotz erhöhter Neurotransmitter-Ausschüttung ein mehr oder weniger
ausgeprägter funktioneller Mangel besteht (Noradrenalin). Serotoninmangel ist ein weiteres
Phänomen der Depression, wobei häufig genetische Faktoren beteiligt sind: verminderte
Synthesekapazität infolge Tph-Polymorphismus oder veränderte Reuptakekinetik durch HHT-
Polymorphismus. Die meisten derzeitigen Antidepressiva basieren auf diesem Phänomen, es
sind SSRI's (Selektive Serotonin-Reuptake Inhibitoren) bzw. NRI's (Noradrenalin-Reuptake-
Inhibitoren) oder kombinierte SNRI's. Wirksame Antidepressiva normalisieren auch die Aktivität
der HVL-NNR-Achse und senken Cortisol.
Für vioele andere Formen der Depression trifft das Hypercortisolismus-Kriterium jedoch nicht zu.
Bei dne reaktiven Depressionen oder „melancholische Depressionen" ist Cortisol vielmehr häufig
erniedrigt, biochemisch nicht zu unterscheiden vom Burnout- oder PTS-Syndrom. Serotonin ist
fast immer niedrig, Noradrenalin normal oder ebenfalls niedrig. Für die Behandlung kommen
heiute neben der reinen selektiven Serotonin-Reuptrakeinhibitoren (SSRI) häufiger Serotonin-
Noradrenalin- (SNRI) oder reine Noradrenalin-Reuptakeinhibitoren (NRI) zum Einsatz. Auch
Störungen der Dopamin-, GABA- und selbst derGlutamatsysteme könneneine Rolle spielen.
S A D / S a i s o n a l A f f e c t i v e D i s o r d e r
Die saisonale Depression (SAD: Saisonal Affective Disorder) kommt vor allem in nördlichen
Ländern vor, wenn sich nach längeren lichtarmen Perioden (Winter) und bei genetischer
Disposition ein ausgeprägter Serotoninmangel entwickeln kann. Heißhungerattacken auf
Kohlehydrate sind bei SAD-Patienten häufig.Lichtmangel beeinträchtigt auf ungeklärte Weise die
Serotoninsynthese. Auch das Vitamin D-Defizit bei fehlendem Sonnenlicht ist beteiligt.
Lichttherapie ist Standard. Eine sehr wirksame Alternative ist die Behandlung mit der
Aminosäurevorstufe 5-HTP oder mit SSRI's bzw. Serotoninsekretagoga wie Fluoxetin.
A d i p o s i t a s
Neben den Hormonen Leptin, Adiponectin, Insulin, HGH, Ghrelin, Glukagon, GLP-1, den
Schilddrüsen- sowie Sexualhormonen sind die Glukokortikoide, CRH, Noradrenalin und vor allem
Serotonin in die Appetitkontrolle und den Energiestoffwechsel involviert. Die Cortisolsekretion ist
bei Übergewichtigen erhöht, bei zentraler (viszeraler) Adipositas stärker als bei peripherer
Adipositas. Serotonin und Noradrenalin sind absolut oder relativ erniedrigt. 10 – 15% der
Adipösen profitieren von alleiniger Behandlung mit Serotoninvorstufen (5-Hydroxytryptrophan)
bzw. selektiven SSRI's, da Serotonin herausragende Bedeutung für die Appetitregulation und
begrenzt auch für den Energiestoffwechsel hat.
Besonders bei Kohlehydrathunger („Craving") sind serotoninerge Strategien sehr wirksam. Reine
Sympathikomimetika wie z.B. Recatol (Phenylpropanolamin) sind in ihrer Wirksamkeit begrenzt.
Sehr effektiv ist die kombinierte Serotonin- und Noradrenalinrestitution mit den betreffenden
Aminosäurevorstufen (5-HTP, Tyrosin) bzw. mit SNRI's, kombinierten Reuptakeinhibitoren wie
Reductil (Sibutramin).
K o p f s c h m e r z e n / M i g r ä n e
Migräne ist eine Erkrankung mit familiärer Häufung, die bis zu 18% der Frauen und 6% der
Männer, vorwiegend im Alter zwischen 30 und 50 Jahren, trifft. Störungen des serotoninergen
Systems sind ursächlich. Kopfschmerz kommt durch Aktivierung des Trigeminussystems
zustande, wobei neuroinflammatorische Peptide wie Substanz P, CGRP (Calcitonin-Gene-
Related Peptide) oder Neurokinine freigesetzt werden. Repetitive neurogene Inflammation
steigert die Erregbarkeit sensorischer Neurone und die Kopfschmerzbereitschaft. Serotonin
hemmt die Schmerzentwicklung über spezifische 5-HT-Rezeptoren auf den Trigeminusfasern.
Bei Migräne liegen genetische Veränderungen der 5HT-Rezptoren vor. Außerdem finden sich
Hinweise für eine immunallergische Komponente. Im akuten Anfall steigt Interleukin 10 an
während IL-4 und IL-5 auch in den Zwischenperioden erhöht sind. Inflammatorische Mediatoren
sind nicht beteiligt, sodass immunologisch ein klares TH2-Aktivitätsmuster dominiert. Auffällig
häufig ist Migräne mit Nahrungsmittelallergien bzw. -unverträglichkeiten assoziiert.
Die Substitution mit den Aminosäurevorstufen 5-HTP und Tyrosin bzw. mit SNRI's ist eine
effektive, spezifische Behandlungsoption.
C o l o n i r r i t a b l e
Funktionelle Magen-Darm-Erkrankungen wie das Reizdarmsyndrom, die funktionelle Dyspepsie
oder funktionelle viszerale Schmerzen haben heute eine Prävalenz von bis zu 20% in den
westlichen Ländern. Viszerale Hypersensitivität und abnorme zentrale Perzeption viszeraler
Signale gelten als Schlüsselelemente der Pathophysiologie. Eine mitentscheidende Rolle spielt
das viszerale neuroendokrine System, zuvorderst Serotonin als Neurotransmitter und parakriner
Mediator. Die enterochromaffinen Zellen des GI-Traktes enthalten ca. 95% des gesamten
Körperserotonins.
Nach vagaler Aktivierung sezernieren enterale Neurone Serotonin, das zur Kontraktion der
glatten Muskulatur, aber auch zur Relaxation im Zusammenspiel mit NO-Neuronen führt. Die
Sekretion wird gesteigert, die Peristaltik vermehrt, wobei verschiedene Serotoninrezeptoren,
insbesondere 5HT1 bis 5HT5, involviert sind. Klinisch können neben den häufigen Schmerzen
sowohl Diarrhoe als auch Obstipation dominieren. Neben Serotoninrezeptor-Agonisten und –
Antagonisten werden vor allem SSRI's eingesetzt.
A D S , A D H S
Das ADS (Aufmerksamkeits-Defizitsyndrom) ist definitionsgemäß eine neurobiologische Störung,
die durch erhebliche Beeinträchtigung der Konzentration und Daueraufmerksamkeit, mangelhafte
Impulskontrolle und eingeschränkte emotionale Regulation gekennzeichnet ist. Bei zusätzlicher
motorischer Hyperaktivität bzw. Unruhe spricht man vom ADHS (ADS mit Hyperaktivität,
„Struwwelpeter-Syndrom"). Folge sind bei Kindern und Heranwachsenden Fehlverhalten in der
Schule, Leistungsschwäche, Lernstörungen, ev. später auch Suchterkrankungen, Depressionen,
Angststörungen. AD(H)S ist eine obligat im Kindesalter beginnende Verhaltens- und Lernstörung,
die jedoch in 30 – 50 % der Fälle auch im Erwachsenenalter fortbesteht. Während die
hyperkinetische Symptomatik oft verschwindet, halten die Aufmerksamkeitsprobleme, die
emotionalen Störungen (Unruhe, Stimmungsschwankungen, Hypersensibilität) und die
Impulsivität an.
Als Ursache des AD(H)S wird heute eine angeborene neurogene Stoffwechselstörung
angesehen, die zur Dysregulation von Neurotransmittersystemen wie Dopamin und Noradrenalin
führt und die geordnete Informationsverarbeitung im Gehirn behindert. Vor allem die
dopaminerge Signaltransduktion ist betroffen. Psychostimulanzien wie das Dopamin-agonistisch
wirkende Ritalin (Methylphenidat), Amphetaminsaft oder Captagon (Fenetyllin) können die
neuronale Aktivität normalisieren. Ritalin wirkt in ca. 70% der Fälle, gelegentliche
ernstzunehmende Nebenwirkungen sind jedoch zu berücksichtigen.
Bei Erwachsenen werden eher tricyclische Antidepressiva (Nortriptylin, Desipramin, Imipramin),
Noradrenalin-Wiederaufnahmehemmer wie Strattera (Atomoxetin) und Edronax (Reboxetin) oder
auch das Antidepressivum Venlafaxin eingesetzt.
Neben der zweifellos vorhandenen genetischen Disposition wird die Rolle verschiedener
Umweltfaktoren kontrovers beurteilt. Reizüberflutung, Rauchen der Mutter in der
Schwangerschaft, familiäre Probleme („schlechtes Elternhaus") sind als aggravierende Faktoren
anerkannt. Schilddrüsenhormone wirken regulierend auf das Neurotransmitternetzwerk.
Subklinische SD-Unterfunktion oder periphere Hormonresistenz werden gehäuft bei AD(H)S-
Kindern gefunden. Unverträglichkeitsreaktionen gegenüber Nahrungsmitteln und NM-Additiva,
Belastung mit Neurotoxinen wie Schwermetallen (Blei, Quecksilber, Aluminium, Cadmium, Arsen)
oder Organochlorverbindungen und Überempfindlichkeit gegenüber Chemikalien (MCS) sind in
ihrer Bedeutung umstritten.
Gesichert ist das gehäufte Vorkommen von Mikronährstoffdefiziten (Magnesium, Zink, Niacin,
Pyridoxin, Thiamin, Folat, Vitamin C, Omega-3-Fettsäuren, seltener auch Vit B12, A, E, B2 und
Pantothenat) bei AD(H)S. Supplementierung mit Vitamin B6, Omega-3-Fettsäuren, Flavonoiden
und Phosphatidylserin scheinen die Symptomatik wesentlich zu verbessern und in Verbindung
mit Detoxifikation, Diät und ggf. Korrektur einer intestinalen Dysbiose äußerst effektiv zu wirken.
Nahrungsmittelunverträglichkeiten sollen bei der Mehrheit der betroffenen Kinder vorkommen.
Eine entsprechende Eliminationsdiät ist daher häufig erfolgreich.
P r ä m e n s t r u e l l e s S y n d r o m
P e r i m e n o p a u s e s y n d r o m
Viele Frauen leiden unter zum Teil erheblichen zyklusabhängigen Beschwerden bzw.
Beschwerden im Rahmen der Menopause: Hitzewallungen, Schmerzen, Unruhe,
Schlafstörungen, Depressionen, Antriebsschwäche, Essstörungen. Neben den hormonellen
Ursachen wie Abfall von Östrogenen, Progesteronmangel, Ungleichgewicht androgener und
östrogener Hormonmengen sind auch neuroregulatorische Defizite mitverantwortlich.
Serotoninmangel, kombiniert mit Noradrenalin- und ev. Dopaminmangel auf der einen Seite,
Defizite inhibitorischer Neurotransmitter wie GABA auf der anderen Seite sind in
unterschiedlichem Maße beteiligt. Behandlung mit Serotonin- bzw. Noradrenalinagonisten
(Clonidin), SSRI's, SNRI's oder auch GABAergegen Substanzen (Gabapentin) sind u.U.
erfolgreicher als der Versuch, hormonelle Defizite auszugleichen.
DIAGNOSTIK
S p e i c h e l h o r m o n e
Die neurohormonelle Funktionsachse wird am besten durch Speichelmessungen von Cortisol
und DHEA geprüft. Speichel hat eine Reihe von Vorteilen gegenüber Blut: die stressfreie
Probengewinnung im normalen Tagesablauf des Patienten; die hohe Stabilität des Testmaterials;
der steilere Tagesgradient der Hormonkonzentrationen. Warum? Im Serum liegen die lipophilen
Steroidhormone wie Cortisol zu 95 - 98% gebunden an Carrierproteine vor (CBG: Cortisol-
bindendes Globulin, Albumin), aktuelle sekretorische Peaks verändern die Hormon-
Gesamtkonzentration im Serum weit weniger als im Speichel, wo ausschließlich das freie
Hormon vorkommt, das mit der freien, biologisch aktiven Hormonfraktion im Serum eng korreliert
ist. Die amerikanische endokrinologische Gesellschaft stuft das Cortisoltagesprofil im Speichel
als optimales Screeningverfahren für Störungen der HPT-HVL-NNR-Achse ein. Die biologische
Psychiatrie kommt nicht mehr ohne Speichelhormonmessung aus.
Bei Schlafstörungen ist das abendliche Cortisol oft erhöht oder steigt sogar später am Abend
nochmals an. In diesen Fällen ist zustzlich zu den 3 Tageswerten auch die spätere Messung von
Cortisol bis 24 Uhr sinnvoll.
DHEA ist zwar im Unterschied zu den anderen Steroidhormonen wie Cortisol durch Sulfatierung
hydrophil und im Blut frei löslich. Im Speichel ist allerdings das nicht-sulfatierte, lipophile DHEA
meßbar, das nur etwa 1% der DHEAS-Konzentration ausmacht aber sehr gut mit DHEAS und
DHEA im Blut korreliert.
N e u r o t r a n s m i t t e r
Für die Bestimmung der Neurotransmitter hat sich der zweite Morgenurin als besonders geeignet
erwiesen. Während die Neurosteroide zyklisch synthetisiert werden und nur morgens zum
Zeitpunkt des Aufstehens ihre Maximalkonzentration erreichen, werden die Neurotransmitter bei
Bedarf ausgeschüttet. Während der erste Morgenurin lediglich die während der nächtlichen Ruhephase abgegebenen NT's enthält, wird im zweiten Morgenurin die normale Tagesaktivität erfasst. Im Unterschied zur konventionellen Diagnostik biogener Amine, bei der die Metaboliten wie die Metanephrine und die Endstufen 5HIES und VMS im Vordergrund stehen, ist die Messung der Neurotransmitter selbst bei der Untersuchung der zentralen Regulation sinnvoll, da sie anders metabolisiert werden. In vielen Arbeiten wurde gezeigt, dass die Neurotransmitter im Urin repräsentativ sind für die Konzentrationen im ZNS (Marc, 2011) und weniger aus dem peripheren Neurotransmitterpool stammen. Dies bestätigen Vergleichsmessungen in Serum, Speichel und Urin, Vergleiche von Urin und Liquor und die therapieabhängigen Veränderungen der NT-Ausscheidung. Die Niere selbst weist starke funktionelle Parallelen zur Synapse auf: sie kann NT's synthetisieren, sezernieren, metabolisieren, wiederaufnehmen und insofern die synaptische Situation repräsentieren. Vor allem Serotonin und Dopamin werden natürlich auch durch deren periphere Quellen beeinflusst. Während die Katecholamine im Blut und Urin korrelieren, ist Serotonin im Serum wesentlich durch das intestinale Serotonin hormonellen Ursprungs geprägt und korreliert nicht mit der Urinausscheidung. Pharmaka können die NT-Ausscheidung maßgeblich beeinflussen, vor allem ß-Blocker, Antihypertensiva und besonders die Antidepressiva. Moderne Antidepressiva sind meist sog. Reuptake-Inhibitoren (SSRI = Selektive Serotonin-Reuptake-Inhibitoren; SNRI = Serotonin-Noradrenalin-Reuptakehemmer; etc.), die quasi am Ende des Problems angreifen. Sie blockieren den Reuptake von NT's am synaptischen Nervenende und erhöhen dadurch die NT-Konzentration an den postsynaptischen Rezeptoren. Dadurch steigern sie allerdings den NT-Verlust, denn die synaptische Wiederverwendung entfällt. Der Reuptake-Hemmeffekt betrifft nicht nur die synaptischen NT-Transporter sondern ebenso natürlich die baugleichen Transporter in BHS, Darmwand, Blutzellen und Nieren. Bei Behandlung mit SSRI oder SNRI ist die Urin-Ausscheidung von Serotonin und ggf. Katecholaminen oft erhöht, obwohl ein zentraler Mangel vorliegen kann. Bei längerer Behandlung kommt es dann infolge wachsenden NT-Defizits häufig zum Absinken der Urinkonzentrationen. Der NeuroStress-Test ist Voraussetzung für die Einleitung einer gezielten individuellen Substitutionsbehandlung. Kontrollen unter Behandlung dienen ggf. die Optimierung der Substitution. Am wichtigsten ist allerdings die gründliche Anamnese mit Feststellung des Beschweredemusters, das Rückschlüsse auf die neuroendokrinen Defizite erlaubt. Zu den häufigsten Ursachen der Dysregulation zählen chronischer beruflicher, sozialer oder psychischer Stress, Insomnie, Schockerlebnisse, chronische Entzündungen, Infektionen oder toxische Belastungen. Nicht selten sind außerdem hormonelle Funktionsstörungen beteiligt. Häufig ist die Schilddrüsenfunktion gestört. Die latente Hypothyreose ist nicht immer sicher am TSH-Anstieg ablesbar und muss durch sorgfältige klinische Beobachtung ausgeschlossen werden; Müdigkeit, Verlangsamung, kognitive Einbußen, Hypotonie, niedrige Körpertemperatur, etc. Auch der vorzeitige oder altersgemäße Abfall der Androgene (Testosteron > DHEA; Testosteron:Östradiol-relation; Progesteron:Östradiol-Quotient) bei Männern und Frauen kann zu ähnlichen Symptomen führen: Kräfteverfall, Müdigkeit, Antriebsschwäche, Muskelabbau, Gewichtszunahme (Fettanteil), Libidoverlust, kognitiven Einbußen, Schlafstörungen und Depressionen. Während DHEA zum Neurostress-Profil gehört, können im Verdachtsfall insbesondere auch Testosteron, Östradiol und Progesteron im Speichel mitgemessen werden. Die Peptidhormone TSH, Prolaktin, ACTH, IGF-1 und auch das Neurosteroid Vitamin D sind nur im Serum/Plasma messbar. Für die Bewertung der Testergebnisse ist besonders wichtig:
1. der Cortisol-Morgenwert, der innerhalb 5 - 10 min nach dem morgendlichen Aufstehen
gewonnen werden muss. Cortisol fällt nach dem Aufstehen rasch auf niedrige Werte ab, sodass bei späterer Entnahme falsch niedrige Werte gefunden werden. Der Morgenwert von Cortisol ist jedoch entscheidend für die Bewertung der HPT-HVL-NNR Achse und die Differenzierung primärer/sekundärer Defekte, Depressionen, Fatigue, CFS, Stress, Burn-Out.
2. Die Cortisol-Verlaufskurve, die u.U. mit deutlichen Anstiegen im Tagesverlauf und ggf.
verlangsamtem/fehlendem Abfall am Abend einhergehen kann. Bei Burn-Out kann der
Morgenwert niedrig und im Tagesverlauf niedrig bleiben oder auch belastungsabhängig ansteigen. Bei primärer Depression ist der Morgenwert hoch und der Abfall zum Abend gering bis fehlend, während bei der sekundären Depression Cortisol morgens erniedrigt ist und im Tagesverlauf stark schwanken kann. Erhöhte Abendwerte korrelieren mit Schlafstörungen.
3. Die Bestimmung der Morgen- und Abendwerte von DHEA liefert wertvolle
Zusatzinformationen über die NNR-Funktion. Der Morgenwert liegt normalerweise 2- bis 5-fach höher als der Abendwert (im Alter abflachend). Ein normaler DHEA-Wert bei niedrigem Cortisol zeigt auf, dass die NNR-Leistungsreserve intakt ist und keine globale NNR-Insuffizienz vorliegt. Bei Cortisolabfall infolge Blockade der HVL-NNR-Achse (Burn-Out, CFS, Fatigue) ist DHEA in der Regel normal oder gar erhöht. Bei lang anhaltendem, chronischem Stress kann auch die DHEA-Synthese betroffen sein und abfallen. In gravierenden Fällen kann der Abend- höher als Morgenwert sein.
4. Die Konzentrationen der einzelnen Neurotransmitter, vor allem das Verhältnis der
exzitatorischen (Noradrenalin, Dopamin, Glutamat, Adrenalin, PEA) zu den inhibitorischen Valenzen (GABA, Serotonin, Glycin) und die Bewertung der einzelnen Neuromodulatoren (Dopamin, Histamin, Taurin oder Glutamin) sind entscheidend für die individuell adaptierte Aminosäuresubstitution. Am häufigsten findet sich in der Praxis ein Mangel an inhibitorischen Neurotransmittern, voran Serotonin, dessen Synthese bei der Mehrzahl der beschriebenen neuroendokrinen Gesundheitsstörungen gehemmt ist. Dabei sind die verschiedenen Möglichkeiten des Serotonindefizits zu bedenken: Ernährungsmängel, Synthesehemmung, entzündliche IDO-Aktivierung, Enzymblockade (Tph), hoher Substratverbrauch oder auch genetische Defekte der Tph oder des SERT.
5. GABA ist bei Hochdruck, chronischen Schmerzen, irritablem Kolon, prämenstruellem
Syndrom, Angststörungen und Depressionen ernieidrigt.
6. Parallel zum Serotoninmangel, findet sich häufig ein Überschuss an Noradrenalin, das
sowohl absolut und mehr noch in Relation zu Adrenalin bei allen akuten/subakuten Stress-assoziierten Störungen ansteigt und bei chronischem Stress zunehmend abfällt. Adrenalin ist häufig stressabhängig erniedrigt, SAMe-Mangel kann ebenfalls Ursache sein. In akuten Konstellationen und bei dominantem Sympathikotonus generell kommt es zum Anstieg.
Dopamin im Urin ist meist unauffällig. Erst bei schweren Regulationsstörungen kann es zum Dopaminmangel – und dann praktisch immer auch zum schweren Noradrenalin- und Adrenalinmangel kommen. Noradrenalin dagegen ist auch bei normalem Dopamin nicht selten mehr oder weniger stark erniedrigt. Offensichtlich ist die Dopaminsynthese weit weniger störungsanfällig als die der nachgeordneten Katecholamine. PEA ist häufig bei Depressionen, Psychosen, chronischer Müdigkeit (CFS) und vor allem bei ADS/ADHS (Aufmerksamkeits-Defizit-Hyperaktivitäts-Syndrom) erniedrigt, bei Stress, Migräne und Schizophrenie erhöht. Bei PEA-Mangel kommt die Substitution mit Phenylalanin in Betracht.
7. Glutamat ist der (mengenmäßig) wichtigste exzitatorische NT, wegen des hohen
Überschusses an Glutamin/GABA jedoch seltener auffällig. Hohe Werte zusammen mit erhöhtem Noradrenalin müssen intensiv mit inhibitorischen NT-Vorstufen behandelt werden (5-HTP, Glutamin, Theanin, etc.).
8. Die Bedeutung von Glycin ist demgegenüber begrenzt. Glycinmangel ist sowohl wegen
dessen Neurotransmitterfunktion als auch wegen der Glutathionsynthese auszugleichen.
9. In seltenen Fällen, bei denen im Vorfeld das Versagen von Antidepressiva bekannt wurde
oder die auf Aminosäuresubstitution atypisch reagieren, kann die molekulargenetische Analyse der wichtigsten Enzympolymorphismen für Serotonin und die anderen Neurotransmitter weiterhelfen. Die Tryptophanhydroxylase vom Typ I oder II ist das limitierende Enzym in der Serotoninsynthese. Genetische Defekte können die Serotoninsynthese um bis zu 80% verringern. An zweiter Stelle ist der Reuptake-Transporter SERT zu nennen, der besonders intensiv untersucht ist. Schließlich die für den Abbau von biogenen Aminen verantwortlichen Enzyme wie COMT (Catechol-O-Methyltransferase) oder MAO (Monoaminoxidase).
10. Schließlich gilt es nochmals zu betonen, dass Störungen der Sexualhormonachse und der
Schilddrüsenfunktion häufig in die Basisdiagnostik einzubeziehen sind, vor allem bei älteren Patienten.
Das Testprogramm für die Feststellung von neuroendokrinen Störungen und die Verlaufskontrolle unter Aminosäure-Substitutionstherapie sollte grundsätzlich nach folgendem Stufenprogramm aufgebaut sein, wobei natürlich individuelle Unterschiede in Symptomatik, klinischer Manifestation, Begleiterkrankungen bzw. –risiken, Vorbehandlung, aktueller Pharmakotherapie zu berücksichtigen sind.
Zeitpunkt 0
Durchführung des Basis-Testprogramms und Behandlungsplanung.
Empfohlenes Basistestprogramm ist das NEUROSTRESS Profil, das neben der
Speichelbestimmung von Cortisol und DHEAS die wichtigsten Neurotransmitter im 2.
MU umfasst: Serotonin, Noradrenalin, Dopamin, Adrenalin, GABA und Glutamat. Eine
kostengünstigere Alternative ist das Profil NEUROSTRESS basis, bei dem die
Messung von GABA und Glutamat im 2. MU wegfällt.
Falls nur mögliche Neurotransmitterstörungen zu untersuchen sind (ADS, ADHS,
Hypotonie, etc) sind die Profile Neurotransmitter I basis mit vier bzw.
Neurotransmitter II plus mit sechs Neurotransmittern im 2. MU sinnvoll.
Wichtige Ergänzungsuntersuchungen zu Behandlungsbeginn sind ggf. DHEAS,
Testosteron und Östradiol im Serum oder auch im Speichel und zusätzlich SHBG,
IGF-1, TSH, fT3 und fT4 im Serum und ACTH im Plasma. Die morgendliche ACTH-
Messung ist in den meisten Fällen geeignet, den Verdacht einer organischen NNR-
Insuffizienz („Adrenal Fatigue") bei niedirgem Speichelcortisol auszuräumen.
Das Testergebnis wird mit ausführlicher Beurteilung und Behandlungsempfehlung
zugeschickt!
nach 2 – 4 Wochen
Behandlungsbeginn auf Basis der vorliegenden Testergebnisse.
In dieser ersten Phase werden unter Berücksichtigung der Basis-Testergebnisse bevorzugt Aminosäurevorstufen inhibitorischer Neurotransmitter wie 5-HTP (5-Hydroxytryptophan; Serotonin), Glutamin (GABA) und die Neuromodulatoren Taurin, Glycin oder Theanin eingesetzt, womit oft schnellere Ergebnisse erzielt werden als bei gleichzeitigem Gabe von inhibitorischen plus exzitatorischen Vorstufen.
Beispiel 1
Serotonin niedrig, Noradrenalin:Adrenalin-Quotient mäßig erhöht, NA normal:
1-2 Kps abends
1- 2 Wochen
anschließend
zusätzlich
Balance D oder DS
1-2 Kps morgens – ev. bis zu 2 – 2 - 0 Kps
Beispiel 2
Serotonin niedrig, NA:A-Quotient hoch, NA hoch, GABA normal - niedrig,
1 – 0 – 2 Kps
2 – 1,2 – 0 Kps
Und bei Schlafstörungen
0 – 0 – 2 Kps abends für 4 - 8 Wochen
Später ev. Umstellung auf
2x2 Kps – Steigerung bis maximal 3 – 3 – 0 Kps
NACplus und GabaNight
0 – 0 - 2 Kps
Beispiel 3
Serotonin niedrig, Dopamin niedrig, NA und A beide niedrig, Glutamat hoch, GABA niedrig. Bei hohem Glutamat ist immer Acetylcystei wichtig, um die Exzitotoxizität von Glutmat zu blockieren.
2 – 1 – 0 Kps bis maximal 3 – 3 – 0 Kps
2 – 1 – 0 Kps, zur Glutamat"entgiftung"
zusätzlich
0 – 0 – 2 Kps
Beispiel 4
Serotonin normal/niedrig, Katecholamine niedrig, Cortisol niedrig. Niedriges Cortisol
wird generell indirekt über Noradrenalinsupport aktiviert, was u.U. längere Zeit in
Anspruch nimmt. Nur in seltenen Fällen kommt die vorübergehende Substitution mit
Hydrocortison in Betracht:
2 – 2 – 0 Kps
Balance DS
2 – 1 – 0 Kps – bis 2 – 2 - 0 Kps
AdreCor
1 – 2 Kps morgens
Neuroendokrine Balance
nach 4–8 Wochen
1. Nach den ersten 6 - 10 Behandlungswochen ev. Laborkontrolle der
Neurotransmitter mit dem Profil Neurotransmitter I oder II und ev.
auch der Speichelsteroide – bei hochpathologischem Erstbefund;
2. Balancierung der Behandlung mit inhibitorischen und verstärkt auch
In dieser Phase werden inhibitorische und exzitatorische Neurotransmitter
gleichermaßen addressiert. Im Zentrum steht daher NeuroReplete mit 2x2-3 Kps tgl.
Diese Phase erstreckt kann sich über mehrere Monate (3 – 6 Monate) erstrecken. Der
aktuelle Bedarf muss regelmäßig in der Praxis an Hand der klinischen Symptomatik
ermittelt werden. Neben NeuroReplete ist AdreCor häufig empfehlenswert, vor allem
bei niedrigen Cortisolwerten zu Beginn.
Kontrolltestung einschließlich der Speichelsteroide wird nach 2 - 4 Monaten
empfohlen, um die Dosierung und die Aminosäurekombinationen anzupassen.
nach 3 - 6 Monaten
Fortsetzung der Behandlung bis zur Wiederherstellung der Gesundheit
Gegenüber Stufe 3 wird nun versucht, die Dosierung der Aminosäurepräparate auf das notwendige Minimum einzustellen. Für die Dauer der Substitutionsbehandlung können keine festen Regeln vorgegeben werden. Sie hängt natürlich vor allem von
den eigentlichen Ursachen der Gesundheitsstörung ab. Handelt es sich – wie sehr häufig - um die Folgen anhaltender, übermäßiger Stressbelastung; handelt es sich um toxische neuronale Schädigung, wobei die Restitution fraglich ist; handelt es sich um ein vorübergehendes Substratdefizit bzw. NT-Synthesehemmung infolge toxischer, alimentärer Einflüsse; in welchem Maße sind genetisch disponierende Faktoren für die Etablierung und Perpetuierung der Störung maßgeblich. Besonders die Restitution der HVL-NNR-Funktionsachse kann längere Zeit in Anspruch nehmen. Relativ schnell kommt die Regeneration der Serotoninspeicher in Gang, dann normalisiert sich Noradrenalin, später erst Adrenalin (und ggf. Dopamin) und häufig erst nach Monaten löst sich die Blockade der Stresshormonachse. Die Herstellung der Balance exzitatorischer und inhibitorischer Signale ist Voraussetzung für die Normalisierung der HVL-NNR-Achse.
Erwartete Veränderungen der Neurotransmitter im zweiten Morgenurin
Nach einigen Tagen
Serotonin erhöht, selten erniedirgt
Nach 2 - 6 Wochen
Noradrenalin, Dopamin, GABA
Nach 3 - 6 Monaten
Adrenalin, Glutamat, NNR-Achse
Anmerkung: Der schnelle Anstieg der Serotoninausscheidung im zweiten Morgenurin korreliert nicht mit der Regeneration der Neurotransmitterpools in den präsynaptischen Vesikeln. Bis zur Stabilisierung der neuronalen Versorgung muss die Substitution länger weitergeführt werden (variiert bei Patienten bis zu mehreren Monaten).
Literatuur
aan het Rot M, Mathew SJ, Charney DS. Neurobiological mechanisms in major depressive disorder. CMAJ 2009;
180(3): 305–313
Adams O, Besken K, Oberdörfer C, McKenzie CR, Takikawa O, Däubener W: Role of indoleamine 2,3-dioxygenase in
alpha/beta and gamma interferon-mediated antiviral effects against herpes simplex virus infections. J Virol 2004; 78:
2632-2636
Agelaki S, Tsatsanis C, Gravanis A, Margioris AN: Corticotropin-releasing hormone augments proinflammatory
cytokine production from macrophages in vitro and in LPS-induced endotoxin shock in mice. Infect Immun 2002; 70:
6068-6074
Agus DB, Gambhir SS, Pardridge WM, Spielholz C, Baselga J, Vera JC, et al. Vitamin C crosses the blood-brain
barrier in the oxidized form through the glucose transporters. J Clin Invest 1997;100:2842–8.
Anisman H. Cascading effects of stressors and inflammatory immune system activation: implications for major
depressive disorder. J Psychiatry Neurosci 2009;34(1):4-20
Arterburn LM, Hall EB, Oken H. Distribution, interconversion, and dose response of n-3 fatty acids in humans. Am J
Clin Nutr 2006; 83(suppl): 1467S – 1476S
Aveldano MI, Bazan NG. Fatty acid composition and level of diacylglycerols and phosphoglycerides in brain and retina.
Biochim Biophys Acta 1973;296(1):1-9
Bangning Y, Becnel J, Zerfaoui M, Rohatgi R, Boulares AH, Nichols CD: Serotonin 5-HT2A receptor activation
suppresses tumor necrosis factor-α-induced inflammation with extraordinary potency. J Pharmacol Exp Ther 2008;
327: 316-323
Barres BA. The mystery and magic of glia: a perspective on their roles in health and disease. Neuron 2008;60(3): 430-40
Beaulieu JM, Zhang X, Rodriguiz RM, Sotnikova TD, Cools MJ, Wetsel WC, Gainetdinov RR, Caron MG. Role of GSK3 beta in behavioral abnormalities induced by serotonin deficiency. Proc Natl Acad Sci U S A. 2008;105(4):1333-8
Berk M, Sanders KM, Pasco JA, Jacka FN, Williams LJ, Hayles AL, Dodd S. Vitamin D deficiency may play a role in depression. Med Hypotheses 2007; 69(6):1316-9. Bethin KE, Vogt SK, Muglia LJ: Interleukin-6 is an essential, corticotropin-releasing hormone-independent stimulator of the adrenal axis during immune system activation. PNAS 2000; 97: 9317-9322
Bierhaus A, Wolf J, Andrassy M, Rohleder N, Humpert PM, Petrov D, Ferstl R, von Eynatten M, Wendt T, Rudofsky G, Joswig M, Morcos M, Schwaninger M, McEwen B, Kirschbaum C, Nawroth PP. A mechanism converting psychosocial stress into mononuclear cell activation. Proc Natl Acad Sci 2003; 100: 1920-25 Binfare RW, Rosa AO, Lobato KR, Santos ARS, Rodrigues ALS. Ascorbic acid administration produces an antidepressant-like effect: evidence for the involvement of monoaminergic neurotransmission. Prog Neuro-Psychopharm Biol Psychiatry 2009; 33: 530-40 Björntorp P, R Rosmond: Obesity and Cortisol. Nutrition 2000; 16: 924-936 Boyer EW, Shannon M: The serotonin syndrome. New Engl J Med 2005; 352: 1112–1120 Bremmer MA, Beekman AT, Deeg DJ, Penninx BW, Dik MG, Hack CE, Hoogendijk WJ. Inflammatory markers in late-life depression: results from a population-based study. J Affect Disord 2008;106(3):249-55 Brody S, Preut R, Schommer K, Schürmeyer TH. A randomized controlled trial of high dose ascorbic acid for reduction of blood pressure, cortisol, and subjective responses to psychological stress. Psychopharmacology 2002;159:319–24 Brotman D, Golden SH, Wittstein IS: The cardiovascular toll of stress. Lancet 2007; 370: 1089-1098 Bryan J. Psychological effects of dietary components of tea: caffeine and L-theanine. Nutr Rev 2008; 66(2):82-90 Calder PC. Glutamine and the immune system. Clin Nutr 1994; 13: 2-8 Calder PC: n-3 Polyunsaturated fatty acids, inflammation, and inflamatory diseases. Am J Clin Nutr 2006; 83(suppl): 1505S-19S Camacho-Arroyo I, López-Griego L, Morales-Montor J. The role of cytokines in the regulation of neurotransmission. Neuroimmunomodulation 2009; 16(1):1-12 Capuron L, Neurauter G, Musselman DL, Lawson DH, Nemeroff CB, Fuchs D, Miller AH. Interferon-alpha-induced changes in tryptophan metabolism. Relationship to depression and paroxetine treatment. Biol Psychiatry 2003; 54: 906–914. Capuron L, Miller AH. Cytokines and psychopathology: lessons from interferon-alpha. Biol Psychiatry 2004; 56: 819–824 Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington HL, McClay J,Mill J, Martin J, Braithwaite A, Poulton R. Influence of Life Stress on Depression: Moderation by a Polymorphism in the 5-HTT Gene. Science 2003; 301: 386-389
Castro MA, Beltrán FA, Brauchi S, Concha II. A metabolic switch in brain: glucose and lactate metabolism modulation by ascorbic acid. J Neurochem. 2009; 110: 423-40 Christiansen L, Tan Q, Iachina M, Bathum L, Kruse TA, McGue M, Christensen K: Candidate Gene Polymorphisms in the Serotonergic Pathway: Influence on Depression Symptomatology in an Elderly Population. Biol Psychiatry 2007;61:223–230 Chrousos GP: Stress, chronic inflammation, and emotional and physical well-being: concurrent effects and chronic sequelae. J Allergy Clin Immunol 2000, 106 (suppl 5): S275-S291 Coenen JJ, Koenen HJ, van Rijssen E, Hilbrands LB, Joosten I: Rapamycin, and not cyclosporin A, preserves the highly suppressive CD27+ subset of human CD4+CD25+ regulatory T cells. Blood 2006; 107: 1018-1023 Contesse V, Lefebvre H, Lengelt S, Kuhn JM, Delarue C, Vaudry H. Role of 5-HT in the regulation of the brain-pituitary-adrenal axis: effects of 5-HT on adrenocortical cells. Can J Physiol Pharmacol 2000 Dec;78(12):967-83 Cowen DS. Serotonin and neuronal growth factors – a convergence of signaling pathways. J Neurochem 2007; 101: 1161-1171 Dagai L, Peri-Naor R, Birk RZ. Docosahexaenoic acid significantly stimulates immediate early response genes and neurite outgrowth. Neurochem Res 2009;34(5):867-75 Dantzer R, O'Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nature Rev Neurosci 2008; 9: 46-57 Das UN. Long-chain polyunsaturated fatty acids in memory formation and consolidation: further evidence and discussion. Nutrition 2003;19(11-12):988-93
Daya S, Anoopkumar-Dukie S : Acetaminophen inhibits liver tryptophan-2,3-dioxygenase activity with a concomitant rse in brain serotonin lvels and a reduction in urinary 5-hydroxyindole acetic acid. Life Sci 2000; 67: 235-240 Dieterich KD, Lehnert H, De Souza EB: Corticotropin-releasing factor receptors: an overview. Exp Clin Endocrinol Diabetes 1997; 105: 65-82 Eisenhofer G, Kopin IJ, Goldstein DS: Catecholamine metabolism: a contemporary view with implications for physiology and medicine. Pharmacol Rev 2004; 56: 331-349 Elenkov IJ, Wilder RL, Chrousos GP, Vizi ES: The sympathetic nerve – an integrative interface between two supersystems: the brain and the immune system. Pharmacol Rev 52: 595-638, 2000 Elenkov IJ, Chrousos GP: Stress System – organization, physiology and immuneoregulation. Neuroimmunomodulation 2006; 13: 257-267 Farooqui AA, Horrocks LA, Farooqui T. Modulation of inflammation in brain: a matter of fat. J Neurochem 2007; 101: 577-99 Fries E, Dettenborn L, Kirschbaum C: The cortisol awakening response (CAR): facts and future directions. Int J Psychophysiology 2008 (epub) Garthwaite J: Neuronal nitric oxide synthase and the serotonin transporter get harmonious. PNAS 2007; 104: 7739-7740 Gerretsen P, Pollock BG. Pharmacogenetics and the serotonin transporter in late-life depression. Expert Opin Drug Metab Toxicol 2008;4(12):1465-78 Gillman PK. Monoamine oxidase inhibitors, opioid analgesics and serotonin toxicity. Br J Anaesth 2005 Oct;95(4):434-41 Golden RN, Heine AD, Ekstrom RD, Bebchuk JM, Leatherman ME, Garbutt JC: A longitudinal study of serotonergic function in depression. Neuropsychopharmacology 2002; 26: 653-659 Grossi G, Perski A, Ekstedt M, Johansson T, Lindström M, Holm K. The morning salivary cortisol response in burnout. J Psychosom Res. 2005 Aug;59(2):103-11 Haghighi F, Bach-Mizrachi H, Huang YY, Arango Y, Shi S, Dwork AJ, Rosoklija G, Sheng HAT, Morozova I, Ju J, Russo JJ, Mann JJ. Genetic architecture of the human tryptophan hydroxylase 2 Gene: existence of neural isoforms and relevance for major depression. Mol Psychiatry 2008 Aug;13(8):813-20 Haskell CF, Kennedy DO, Milne AL, Wesnes KA, Scholey AB. The effects of L-theanine, caffeine and their combination on cognition and mood. Biol Psychol 2008;77(2):113-22 Hauser P, Khosla J, Aurora H, Laurin J, Kling MA, Hill J, Gulati M, Thornton AJ, Schultz RL, Valentine AD, Meyers CA, Howell CD. A prospective study of the incidence and open-label treatment of interferon-induced major depressive disorder in patients with hepatitis C. Mol Psychiatry 2002; 7: 942–947 Hellhammer DH, Wüst S, Kudielka BM: Salivary cortisol as a biomarker in stress research. Psychoneuroendocrinology 2008 (epub) Herman JP, Mueller NK, Figueiredo H. Role of GABA and Glutamate Circuitry in Hypothalamo-Pituitary-Adrenocortical Stress Integration. An NY Acad Sci 1018: 35-45, 2004
Hoogendijk WJ, Lips P, Dik MG, Deeg DJ, Beekman AT, Penninx BW. Depression is associated with decreased 25-hydroxyvitamin D and increased parathyroid hormone levels in older adults. Arch Gen Psychiatry. 2008; 65(5):508-12 Hoffmann A, Levchenko, Scott ML, Baltimore D: The IkB – NF-kB signaling module: temporal control and selective gene activation. Science 298: 1241-1245, 2002
Imeri L, Opp MR. How (and why) the immune system makes us sleep. Nat Rev Neurosci 2009;10(3):199-210
Jorde R, Waterloo K, Saleh F, Haug E, Svartberg J. Neuropsychological function in relation to serum parathyroid hormone and serum 25-hydroxyvitamin D levels. The Tromsoe study. J Neurol 2006; 253: 464-470 Jorgensen HS. Studies on the neuroendocrine role of serotonin. Dan Med Bull 2007; 54: 266-288 Kai S, Goto S, Tahara K, Sasaki A, Kawano K, Kitano S: Inhibition of indoleamine 2,3-dioxygenase suppresses NK cell activity and accelerates tumor growth. J Exp Ther Oncol 2003; 3: 336-45 Kaufman RE, Ostacher MJ, Marks EH, Simon NM, Sachs GS, Jensen JE, Renshaw PE, Pollack MH. Brain GABA levels in patients with bipolar disorder. Progr Neuro-Psychopharm Biol Psychiatry 33 (2009) 427–434
Kim H, Lim SW, Kim SW, Kim JW, Chang YH, Carroll BJ, Kim DK. Monoamine transporter gene polymorphisms and antidepressant response in koreans with late-life depression. JAMA 2006;296(13):1609-18 Kimura K, Ozeki M, Juneja LR, Ohira H. L-Theanine reduces psychological and physiological stress responses. Biol Psychol. 2007; 74(1):39-45. Kirby ED, Geraghty AC, Ubuka T, Bentley GE, Kaufer D. Stress increases putative gonadotropin inhibitory hormone and decreases luteinizing hormone in male rats. PNAS 2009; (epub June 18) Kirchheiner J, Henckel HB, Franke L, Meineke I, Tzvetkov M, Uebelhack R, Roots I, Brockmöller J. Impact of the CYP2D6 ultra-rapid metabolizer genotype on doxepin pharmacokinetics and serotonin in platelets. Pharmacogenetics and Genomics 2005; 15: 579–587 Kolenda KD: Secondary prevention of coronary heart disease. How effective are modern therapy methods ? Dtsch Med Wochenschr 2003; 128: 1847-48 Koo JW, RS Duman. IL-1ß is an essential mediator of the antineurogenic and anhedonic effects of stress. PNAS 2008; 105: 751-56 Kraft JB, Peters EJ, Slager SL, Jenkins GD, Reinalda MS, McGrath PJ, Hamilton SP. Analysis of association between serotonin transporter and antidepressant response in a large clinical sample. Biol Psychiatry 2007; 61: 734-742 Kudielka BM, Hellhammer DH, Wüst S: Why do we respond so differently? Reviewing determinants of human salivary cortisol responses to challenge. Psychoneuroendocrinology 2009; 34: 2-18
Lee CK, Weindruch R, Prolla TA. Gene-expression profile of the ageing brain in mice. Nat Genet 2000; 25: 294-97 Levy HL: Metabolic disorders in the center of genetic medicine. N Engl J Med 2005;353: 1968-1970
Lukiw WJ, Bazan NG. Docosahexaenoic acid and the aging brain. J Nutr 2008: 138: 2510-14
Lupien SJ, McEwen BS, Gunnar MR, Heim C. Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nat Rev Neurosci. 2009 Jun;10(6):434-45 Yusuf S, Hawken S, Ounpuu S, Dans T, Avezum A, Lanas F, McQueen M, Budaj A, Pais P, Varigos J, Lisheng L; INTERHEART STUDY Investigators: Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): a case control study. Lancet 2004; 364: 937-52 Manninger N, Wolkowitz OM, Reus VI, Epel ES, Mellon SH: Neurobiological and Neuropsychiatric Effects of Dehydroepiandrosterone (DHEA) and DHEA Sulfate (DHEAS). Front Neuroendocrinol 2008 (epub) Marsland AL, Gianaros PJ, Abramowitch SM, Manuck SB, Hariri AR: Interleukin-6 covaries inversely with hippocampal grey matter volume in middle-aged adults. Biol Psychiatry 2008; 64: 484-490 Marc DT, Ailts JW, Ailts Campeau DC, Bull MJ, Olson KL. Neurotransmitters excreted in the urine as biomarkers of nervous system activity: Validity and clinical applicability. Neurosci Biobehav Rev 2011; 35: 635-644 Marz P, Cheng JP, Gadient RA, Patterson PH, Stoyan T, Otten U, Rose-John S: Sympathetic neurons can produce and respond to interleukin 6. PNAS 1998; 95: 3251-3256 Mathe G: The need of a physiologic and pathophysiologic definition of stress. Biomed Pharmacother 2000; 54: 119-121
McAfoose J, Baune BT. Evidence for a cytokine model of cognitive function. Neurosci Biobehav Rev 2009 Mar;33(3):355-66
McEwen BS. Physiology and neurobiology of stress and adaptation: central role of the brain. Physiol Rev 2007;87(3):873-904
McEwen BS. Central effects of stress hormones in health and disease: Understanding the protective and damaging effects of stress and stress mediators. Eur J Pharmacol. 2008; 583(2-3):174-85 McMahon FJ, Buervenich S, Charney D, Lipsky R, Rush AJ, Wilson AF, Sorant AJ, Papanicolaou GJ, Laje G, Fava M, Trivedi MH, Wisniewski SR, Manji H. Variation in the gene encoding the serotonin 2A receptor is associated with outcome of antidepressant treatment. Am J Hum Gen 2006; 78(5): 804-14 Miller AH. Therapeutic considerations of L-glutamine: a review of the literature. Altern Med Rev 1999; 4: Miller AH, Ancoli-Israel S, Bower JE, Capuron L, Irwin MR: Neuroendocrine-Immune Mechanisms of Behavioral Comorbidities in Patients With Cancer. J Clin Oncol 2008; 26:971-982
Miller GE, Chen E, Sze J, Matin T, Arevalo JM, Doll R, Ma R, Cole SW: A functional genomic fingerprint of chronic stress in humans: blunted glucocorticoid and increased NF-kappaB signaling. Biol Psychiatry 2008; 64: 266-272
Miller AH, Maletic V, Raison CL. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry. 2009 May 1;65(9):732-41 Müller N, Riedel M, Schwarz MJ, Engel RR: Clinical effects of COX2 inhibitors on cognition in schizophrenia. Eur Arch Psychiatry clin Neurosci 2005; 255: 149-151 Müller N, Schwarz MJ: The immune-mediated alteration of serotonin and glutamate: towards an integrated view of depression. Mol Psychiatry 2007 12, 988–1000 Müller N, Schwarz MJ. COX-2 inhibition in schizophrenia and major depression. Curr Pharm Des. 2008;14(14):1452-65 Müller N, Schwarz MJ: A psychoneuroimmunological perspective to Emil Kraepelins dichotomy. Eur Arch Psychiatry Clin Neurosci 2008; 258 (suppl 2): 97-106 Munn DJ, Shifizadeh E, Attwood JT, Bondarev I, Pashine A, Mellor AL: Inhibition of T cell proliferation by macrophage tryptophan catabolism. J Exp Med 189: 1363-1372, 1999 Myint AM, Kim YK. Cytokine-serotonin interaction through IDO: a neurodegeneration hypothesis of depression. Med Hypotheses 2003; 61: 519–525
Myint AM, Schwarz MJ, Steinbusch HW, Leonard BE. Neuropsychiatric disorders related to interferon and interleukins treatment. Metab Brain Dis. 2009; 24(1):55-68. Nater UM, Maloney E, Boneva RS, Gurbaxani BM, Lin JM, Jones JF, Reeves WC, Heim C. Attenuated morning salivary cortisol concentrations in a population based study of persons with chronic fatigue syndrome and wel controls. J Clin Endocrinol Metab 2008; 93(3): 703-709 Nautiyal KM, Ribeiro AC, Pfaff DW, Silver R: Brain mast cells link the immune system to anxiety-like behavior. PNAS 2008: 105: 18053-57 Newsholme EA, Blomstrand E. Branched-chain amino acids and central fatigue. J Nutr 2006; 136 (suppl 1): 274S-76S Nichols DE, Nihols CD: Serotonin receptors. Chem Rev 2008; 108: 1614-41 Nishizawa S, Benkelfat C, Young SN, Leyton M, Mzengeza S, de Montigny C, Blier P, Diksic M : Differences between males and females in rates of serotonin synthesis in human brain. PNAS 1997; 94, 5308-5313 Ohtsuki S: New Aspects of the Blood–Brain Barrier Transporters; Its Physiological Roles in the Central Nervous System. Biol. Pharm Bull 2004; 27(10) 1489—1496 Osterberg K, Karlson B, Hansen AM. Cognitive performance in patients with burnout, in relation to diurnal salivary cortisol. Stress 2009; 12(1): 70-81 Pecchi E, Dallaporta M, Jean A, Thirion S, Troadec JD. Prostaglandins and sickness behavior: old story, new insights. Physiol Behav 2009; 97: 279-92 Pepine CJ, Raczynski JM, Light K, Krantz DS, Stone PH, Knatterud GL, Kaufmann PG: Mental stress-induced ischemia and all-cause mortality in patients with coronary artery disease: results from the Psychophsiological Investigatons of Myocardial Ischemia study. Circulation 2002; 105: 1780-1784 Pérez-Neri I, Montes S, Ojeda-López C, Ramírez-Bermúdez J, Ríos C: Modulation of neurotransmitter systems by dehydroepiandrosterone and dehydroepiandrosterone sulfate: Mechanism of action and relevance to psychiatric disorders Progr NeuroPsychopharmacol Biol Psychiatry; 2008; 32: 1118–1130 Perianayagam MC, Oxenkrug GF, Jaber BL: Immune-modulating effects of melatonin, N-acetylserotonin, and N-acetyldopamine. Ann NY Acad Sci 2005; 1053: 386–393 Perlis RH, Fijal B, Adams DH, Sutton VK, Trivedi MH, Houston JP. Variation in Catechol-O-Methyltransferase is associated with duloxetine response in a <clinical trial for major depressive disorder. Biol Psychiatry 2009;65: 785-791 Peters EJ, Slager SL, McGrath PJ, Knowles JA, Hamilton SP. Investigation of serotonin-related genes in antidepressant Response. Molecular Psychiatry (2004) 9, 879–889 Petersen AMW, Pedersen BK: The role of IL-6 in mediating the antiinflammatory effects of exercise. J Physiol Pharmacol 2006; 57 (suppl 10): 43-51
Petty F. Plasma concentrations of gamma-aminobutyric acid (GABA) and mood disorders: a blood test for manic depressive disease? Clin Chem 1994; 40(2):296-302 Raison CL, Borisov AS, Majer M, Drake DF, Pagnoni G, Woolwine BJ, Vogt GJ, Massung B, Miller AH. Activation of central nervous system inflammatory pathways by interferon-alpha: relationship to monoamines and depression. Biol Psychiatry. 2009 Feb 15;65(4):296-303
Raison CL, Borisov AS, Woolwine BJ, Massung B, Vogt G, Miller AH. Interferon-alpha effects on diurnal hypothalamic-pituitary-adrenal axis activity: relationship with proinflammatory cytokines and behavior. Mol Psychiatry. 2008 Jun 3 [Epub ahead of print] Refojo D, Liberman AC, Holsboer F, Arzt F. Transcription factor-mediated molecular mechanisms involved in the functional cross-talk between cytokines and glucocorticoids. Immunol Cell Biol 79: 385-394, 2001 Rogers PJ, Smith JE, Heatherley SV, Pleydell-Pearce CW. Time for tea: mood, blood pressure and cognitive performance effects of caffeine and theanine administered alone and together. Psychopharmacology 2008; 195(4):569-77 Rotondo A, Mozzanti C, Dell'Osso L, Rucci P, Sullivan P, Bouanani S, Gonnelli C, Goldman D, Cassano GB. Catechol O-Methyltransferase, Serotonin Transporter, and Tryptophan Hydroxylase Gene Polymorphisms in Bipolar Disorder Patients With and Without Comorbid Panic Disorder. Am J Psychiatry 2002 159: 23-29
Rupp H, Wagner D, Rupp T, Schulte LM, Maisch B. Risk stratification by the "EPA+DHA level" and the "EPA/AA ratio" focus on anti-inflammatory and antiarrhythmogenic effects of long-chain omega-3 fatty acids. Herz 2004;29(7):673-85. Sanacora G, Rothman DL, Mason G, Krystal JH: Clinical studies implementing glutamate neurotransmission in mood disorders. Ann NY Acad Sci 2003; 1003: 292–308 Schiepers OJ, Wichers MC, Maes M: Cytokines and major depression. Prog Neuropsychopharmacol Biol Psychiatry 2005; 29: 201-217 Schwieler L, Erhardt S, Erhardt G, Engberg G: Prostaglandin-mediated control of rat brain kynurenic acid synthesis – opposite actions by COX1 and COX2 isoforms. J Neural Transm 2005; 112: 863-872
Sei Y, Vitkoviç L, Yokoyama MM. Cytokines in the Central Nervous System: Regulatory Roles in Neuronal Function, Cell Death and Repair. Neuroimmunomodulation 1995;2:121-133
Sheps DS, McMahon RP, Becker L, Carney RM, Freedland KE, Cohen JD, Sheffield D, Goldberg AD, Ketterer MW,
Shonkoff JP, Boyce WT, McEwen BS. Neuroscience, molecular biology, and the childhood roots of health disparities:
building a new framework for health promotion and disease prevention. JAMA. 2009 Jun 3;301(21):2252-9
Smits KM, Smits LJ, Schouten JS, Peters FP, Prins MH. Does pretreatment testing for serotonin transporter
polymorphisms lead to earlier effects of drug treatment in patients with major depression? A decision-analytic model.
Clin Ther 2007; 29(4): 691-702
Su S, Snieder H, Miller AH, Ritchie J, Bremner JD, Goldberg J, Dai J, Jones L, Murrah NV, Zhao J, Vaccarino V.
Genetic and environmental influences on systemic markers of inflammation in middle-aged male twins. Atherosclerosis.
2008 Sep;200(1):213-20
Sun-Edelstein C, Tepper SJ, Shapiro REDrug-induced serotonin syndrome: a review. Expert Opin Drug Saf. 2008
Sep;7(5):587-96
Szelenyi J, Vizi ES: The catecholamine cytokine balance: interaction between the brain and the immune system. Ann .
Aca. Sc.2007; 1113: 311–324
Tracey KJ: Physiology and immunology of the cholinergic antiinflammatory pathway. J Clin Invest 2007; 117: 289-296
Tsai SJ, Hong CJ, Liou YJ, Yu YWJ, Chen TJ, Hou SJ, Yen FC. Tryptophan hydroxylase 2 gene is associated with
major depression and antidepressant treatment response. Prog Neuro-Psychopharmacol Biol Psychiatry 2009 (4),
epub
Vaccarino V, Brennan ML, Miller AH, Bremner D, Ritchie JC, Lindau F, Veledar E, Su S, Murrah NV, Jones L, Jawed F,
Dai J, Goldberg J, Hazen SL: Association of major depressive disorder with serum myeloperoxidase and other markers
of inflammation: a twin study. Biol Psychiatry 2008; 64: 476-483
Vgontzas AN, Bixler EO, Lin HM, Prolo P, Trakada G, Chrousos GP. IL-6 and its circadian secretion in humans.
Neuroimmunomodulation 2005; 12: 131-40
Vielhaber K, Riemann D, Feige B, Kuelz A, Kirschbaum C, Voderholzer U: Impact of experimentally induced serotonin deficiency by tryptophan depletion on saliva cortisol concentrations. Pharmacopsychiatry 2005; 38: 87-94 Walther DJ, Peter JU, Bashammakh S, Hörtnagl H, Voits M, Fink H, Bader M. Synthesis of Serotonin by a Second Tryptophan Hydroxylase Isoform. Science 2003; 299: 76 Weaver KL, Ivester P, Seeds M, Case LD, Arm JP, Chilton FH. Effect of dietary fatty acids on inflammatory gene expression in healthy humans. J Biol Chem 2009; 284: 15400-407 Whang W, Kubzansky LD, Kawachi I, Rexrode KM, Kroenke CH, Glynn RJ, Garan H, Albert CM. Depression and risk of sudden cardiac death and coronary heart disease in women. J Am Coll Cardiol 2009; 53: 950-58 Wilbert-Lampen U, Straube F, Trapp A, Deutschmann A, Plasse A, Steinbeck G: Effects of corticotropin-releasing hormone (CRH) on monocyte function, mediated by CRH-receptor subtype R1 and R2. J Cardiovasc Pharmacol 2006; 47: 110-116
Wilkins CH, Sheline YI, Roe CM, Birge SJ, Morris JC. Vitamin D deficiency is associated with low mood and worse
cognitive performance in older adults. Am J Geriatr Psychiatry. 2006; 14(12):1032-40
Wilson CJ, Finch CE, Cohen HJ. Cytokines and Cognition - The Case for A Head-to-Toe Inflammatory Paradigm. J Am
Ger Soc 2002; 50:2041–2056
Wurtman RJ: Genes, stress, and depression. 2005;54(5 Suppl 1):16-9
Yu A, Idle JR, Byrd LG, Krausz CW, Küpfer A, Gonzalez FJ. Regeneration of serotonin from 5-methoxytryptamine by
polymorphic human CYP2D6. Pharmacogenetics 2003, 13:173–181
Zeisel SH, Blusztajn JK: Choline and human nurition. Annu Rev Nutr 1994; 14: 269-296
Zhang X, Beaulieu JM, Sotnikova TD, Gainetdinov RR, Caron MG. Tryptophan Hydroxylase-2 Controls Brain Serotonin
Synthesis. Science 2004; 305: 217
Zlokovic BV: The Blood-Brain Barrier in Health and Chronic Neurodegenerative Disorders. Neuron 2008: 57: 178-201
Zoumakis E, Kalantaridou SN, Chrousos GP: The "brain-skin connection": nerve growth-factor-dependent pathways for
stress-induced skin disorders. J Mol Med 2007; 85: 1347-49
Neurotransmitter - Platelets
Serotonin
Reduced uptake – MDD Meltzer HY, Arora RC, Baber R, Tricou BJ. Arch Gen Psychiatry. 1981 Dec;38(12):1322-6
Meltzer HY, Arora RC. Genetic control of serotonin uptake in blood platelets: a twin study. Psychiatry Res. 1988 Jun;24(3):263-9.
Greenberg BD, Tolliver TJ, Huang SJ, Li Q, Bengel D, Murphy DL. Genetic variation in the serotonin transporter promoter region affects serotonin uptake in human blood platelets. Am J Med Genet. 1999 Feb 5;88(1):83-7 Javors MA, Seneviratne C, Roache JD, Ait-Daoud N, Bergeson SE, Walss-Bass MC, Akhtar FZ, Johnson BAPlatelet serotonin uptake and paroxetine binding among allelic genotypes of the serotonin transporter in alcoholics. Prog Neuropsychopharmacol Biol Psychiatry. 2005 Jan;29(1):7-13
Source: http://dr-bieger.de/wp-content/uploads/2011/10/Dr_Bieger-NeuroStress_Guide-Juli.2009-kurz2011.pdf
Information for Patients ROTATOR CUFF TEARS This booklet has been prepared to help you better understand your shoulder problem and the surgery that may be required. It will also help explain what will happen after the surgery has been performed. Although this booklet aims to be relatively comprehensive, you will probably still have some questions and I would of course be happy to answer these at any time.
Improved Al ograft Disinfection Technique Using GCIB Joseph Khoury, PhD1; Sean R Kirkpatrick1; Son Chau1; Raymond Cherian1; Cameron McCaa1,2; Richard C Svrluga1; Laurence JB Tarrant, PhD1 INNOVATIONS IN SURFACE PROCESSING Exogenesis Corp. Billerica, MA 01821 2McCaa Group, Atlanta, GA 30307 Materials and Methods Background: Currently, soft tissue al ografts are classified in two broad groups; aseptical y harvested or