Microsoft word - dhaen_compiled.doc
Neuroendocrinology
David A. Gutman and Charles B. Nemeroff
For publication in: Textbook of Biological Psychiatry
The occurrence of psychiatric symptoms such as thought disturbances and depressed
mood in patients with primary endocrine disorders is common; in addition a significant
percentage of patients with psychiatric disorders demonstrate a consistent pattern of endocrine
dysfunction. Our understanding of the neurobiology of depression and other psychiatric
disorders has been aided tremendously by a systematic analysis of the neuroendocrine axes and
the actions of neurohormones in the pituitary gland and throughout the central nervous system
(CNS). The occurrence of prominent psychiatric symptoms in patients with primary endocrine
disorders including Cushing's disease and primary hypothyroidism provided a rationale for
exploring the connection between hormones and both affective and cognitive function. In fact,
disorders of neuroendocrine dysregulation in subpopulations of psychiatric patients are among
the most consistent neurobiological findings in all of biological psychiatry.
Bleuler was among the earliest investigators to systematically investigate the association
between hormones, mood and behavior. He first demonstrated that patients with primary
endocrine disorders have higher than expected psychiatric morbidity, which often resolved after
correcting the primary hormonal abnormality. Work over the past 25 years has clearly
demonstrated that the CNS tightly regulates endocrine gland secretion, and further, that neurons
are directly influenced by hormones.
The concept that neurons are capable of synthesizing and releasing hormones initially
sparked a controversy in endocrinology and neuroscience when first introduced in the 1950s;
namely, is it possible that certain neurons subserve endocrine functions? Two major findings
fueled this debate. First, neurohistologists working with mammalian, as well as lower vertebrate
and invertebrate species made several key observations. Led by a husband and wife team, the
Scharrers, early researchers documented, by both light and electron microscopy, the presence of
neurons that had all the characteristics of previously studied endocrine cells. These neurons
stained positive with the Gomori stain, which was believed to be specific to endocrine tissues,
and further they contained granules or vesicles containing known endocrine substances. The
second key area of research centered around the brain's control of the secretion of pituitary
trophic hormones. These trophic hormones were long known to control the secretion of
peripheral target endocrine hormones, e.g. thyroid hormone, gonadal steroids, adrenal steroids,
etc. These interactions were particularly compelling because of the earlier identification of an
extremely important neuroendocrine system, namely the magnocellular cells of the
paraventricular nucleus (PVN) of the hypothalamus, which synthesize vasopressin and oxytocin.
These two nonapeptides were shown to be transported from PVN cell bodies down the axon to
nerve terminals located in the posterior pituitary (neurohypophysis), and released in response to
appropriate physiologic stimuli. Vasopressin, also known as antidiuretic hormone, is a critical
regulator of fluid balance, and oxytocin regulates the milk-letdown reflex during breast-feeding.
The ability of neurons to function as true endocrine tissues has now been clearly
established. Neural tissue can both synthesize and release substances, known as
(neuro)hormones, that are released directly into the circulatory system, and have effects at sites
far removed from the brain. One important example noted above is the action of vasopressin on
the kidney. Although early in the development of the emerging discipline of neuroendocrinology
it seemed important to document the ability of neurons to function as neuroendocrine cells,
particularly those in the CNS, classification of specific chemical messengers as either endocrine
versus neuronal versus neuroendocrine soon lost its heuristic value. It is now recognized that the
same substance can act as a neurotransmitter and a hormone depending on its location within the
CNS and periphery. A good example of this is epinephrine (adrenaline), which functions as a
classical hormone in the adrenal medulla but as a conventional neurotransmitter in the
mammalian CNS. Similarly it has been demonstrated that corticotropin-releasing factor (CRF)
functions as a true peptide hormone in its role as a hypothalamic hypophysiotropic factor in
promoting the release of adrenocorticotropin (ACTH) from the anterior pituitary, yet also
functions as a ‘conventional' neurotransmitter in cortical and limbic areas. Thus the field now
seeks to elucidate the role of particular chemical messengers in particular brain regions or
The traditional endocrine and hormonal functions for several peptides discussed above
have been well established, but many of these substances may also possess paracrine roles as
well, i.e. secretion of these substances from one cell acts upon proximal cells. These paracrine
interactions remain largely unexplored. The importance of these paracrine effects has been well
demonstrated in the gastrointestinal tract where several peptides that act as hormones or
neurotransmitter substances at other sites, including the CNS, have influences on local cellular
function. Examples include vasoactive intestinal peptide, cholecystokinin and somatostatin.
Overview of components and control mechanisms
The hypothalamic-pituitary-end-organ axes generally are organized in an hierarchical
fashion [Figure 1]. A large percentage of the neuroendocrine abnormalities in patients with
psychiatric disorders are related to disturbances of target hormone feedback. A generic
description is briefly outlined here. More comprehensive reviews on this topic are available
(Levine 2000). In general, the hypothalamus contains neurons that synthesize and release factors
that either promote or inhibit the release of anterior pituitary hormones, so-called release or
release-inhibiting factors. These peptide hormones, as summarized in Table 1, are synthesized
by transcription of the DNA sequence for the peptide prohormone. After translation in the
endoplasmic reticulum, these prohormones are processed during axonal transport and packaged
into vesicles destined for the nerve terminals. These now biologically active peptides are then
released following appropriate physiological stimuli from the median eminence, the most ventral
portion of the hypothalamus, and secreted into the primary plexus of the hypothalamo-
hypophyseal portal vessels [Figure 2]. These peptides are transported in high concentration to
the sinusoids of the anterior pituitary (adenohypophysis) where they bind to specific membrane
receptors on their targets, the pituitary trophic-hormone producing cells. Activation of these
receptors promotes or inhibits the release of pituitary trophic hormones into the systemic
circulation. The increase or decrease in the plasma concentrations of these pituitary trophic
hormones produces a corresponding increase or decrease in their respective end-organ hormone
secretion. The hormones of the end-organ axes, such as gonadal and adrenal steroids, feedback
on both pituitary and hypothalamic cells to prevent further release, often referred to as "long-
loop" negative feedback. Short-loop negative feedback circuits have also been identified in
which pituitary hormones directly feedback on hypothalamic neurons to prevent further release
of hypothalamic releasing factors.
Disturbances in the feedback regulation of the hypothalamic-pituitary-end organ axes are
of considerable interest in psychiatry. The common occurrence of psychiatric symptoms in
many primary endocrine disorders, such as hypothyroidism and Cushing's syndrome, served as
an impetus for investigation into the regulation of neuroendocrine systems in psychiatric disease
states such as depression, schizophrenia and bipolar disorder. Thus, a large part of
psychoneuroendocrinology has focused on identifying changes in basal levels of pituitary and
end-organ hormones in patients with psychiatric disorders. For many of the axes discussed
below, tests have been developed to assess the functional status of these feedback systems. In
these so-called stimulation tests, hypothalamic and/or pituitary derived factors or their synthetic
analogs are exogenously administered, and the hormonal response to this "challenge" is assessed.
For example, in the standard corticotropin-releasing-factor (CRF) stimulation test, a 1 μg/kg dose
of CRF is administered intravenously, and the adrenocorticotropin (ACTH) and cortisol response
is measured over a period of 2 or 3 hours. This test is a very sensitive measure of hypothalamic-
pituitary-adrenal (HPA) axis activity, and changes in the magnitude and/or duration of the
response relative to normal control values are characteristic of one or another type of
dysregulation of the HPA axis.
Limitations of stimulation tests:
Such studies as outlined above provide valuable information, but a brief discussion of some
inherent limitations is warranted before a detailed review of the literature is presented. Normal
circadian rhythms and the pulsatile release of many of the hypothalamic-pituitary-end-organ axes
components are often not taken into account when these stimulation tests are designed. Further,
differences in assay sensitivity, gender differences, inclusion criteria for patients used in studies,
and severity of symptoms in the target patient population studied can potentially generate
confounding or at least quite variable results. Nevertheless, a great deal about the neurobiology
of psychiatric disorders has been discovered through such experiments.
Although less commonly used today, an often-utilized strategy in the 1970s and 1980s was
based on the perception that the neuroendocrine axes served as a "window" into CNS function.
Peripheral neuroendocrine markers were often used to indirectly assess CNS function because
the brain was relatively inaccessible for study, with the exception of cerebrospinal fluid (CSF)
and postmortem studies. With the emergence of the monoamine theories of mood disorders and
schizophrenia, many investigators attempted to draw conclusions about the activity of
noradrenergic, serotonergic, and dopaminergic circuits in patients with various psychiatric
disorders by measuring the basal and stimulated secretion of pituitary and end-organ hormones
in plasma. Although these approaches have severe limitations, they have been useful in
elucidating the pathophysiology of mood and anxiety disorders, and to a lesser extent,
In summary neuroendocrinology broadly encompasses the following:
♦ The neural regulation of the secretion of peripheral, target-organ hormones, pituitary
trophic hormones, and hypothalamic-hypophysiotropic hormones.
♦ The effects of each of the hormones that comprise the various endocrine axes on the
CNS. This includes, for example, the effects of synthetic glucocorticoids on memory
♦ Study of alterations in the activity of the various endocrine axes in major psychiatric
disorders, and conversely the behavioral consequences of endocrinopathies.
Dysregulation of the hypothalamic-pituitary-adrenal (HPA) axis has frequently been
reported in patients with psychiatric disorders and is among the most robustly demonstrated
neurobiological changes among psychiatric patients. The primary regulator of this axis is
corticotropin-releasing factor (CRF), also known as corticotropin-releasing hormone (CRH), a 41
amino acid containing peptide synthesized in parvocellular neurons located primarily in the
paraventricular nucleus (PVN) of the hypothalamus. CRF containing cells in the PVN receive
input from a variety of brain nuclei including the amygdala, bed nucleus of the stria terminalis,
and other brain stem nuclei (Hauger 2000). These CRF-containing neurons in turn project to
nerve terminals in the median eminence (Swanson, et al. 1983), and CRF is released into the
hypophyseal-portal system where it activates CRF receptors on corticotrophs in the anterior
pituitary to promote the synthesis of pro-opiomelanocortin (POMC) and the release of its post-
translational products, adrenocorticotropin hormone (ACTH), β-endorphin and others [Figure 4].
Arginine-vasopressin (AVP) also promotes the release of ACTH from the anterior pituitary,
though CRF is necessary for AVP to exert this effect. Chronic stress can also upregulate AVP
expression in the PVN, where under these conditions it may be coexpressed in CRF containing
neurons (Hauger 2000). ACTH released from the anterior pituitary in turn stimulates the
production and release of cortisol, the primary glucocorticoid in humans, from the adrenal cortex
The concentration of circulating glucocorticoids is modulated via long-loop negative
feedback. An increase in circulating glucocorticoids inhibits hypothalamic CRF gene expression
and ACTH secretion from the pituitary. This in turn prevents further glucocorticoid release. The
HPA axis also undergoes a circadian rhythmicity in humans where serum cortisol levels peaks
before immediately before awakening and reaches is nadir in the evening.
The biologic effects of glucocorticoids are regulated by two cytosolic receptors: the
glucocorticoid receptor (GR) or the mineralocorticoid receptor (MR), which both belong to a
large superfamily of steroid hormone receptors. Because the mineralocorticoid receptor has a
much higher affinity for glucocorticoids than does the glucocorticoid receptor, MR binding sites
may be saturated with glucocorticoids under physiological conditions. In contrast, the
occupancy of GR binding sites change in response to changes in circulating glucocorticoid
levels. The main genomic affects of glucocorticoids are mediated by GR binding to
glucocorticoid response elements (GREs) in the promoter regions of specific genes. GRs may
also inhibit or enhance the actions of other transcription factors such as AP-1, NF-ĸB, and
CREB, by direct protein-protein interactions (Nestler 2001).
The biology of Corticotropin-Releasing Factor (CRF)
Although Saffron and Schally identified a crude extract which promoted the release of
ACTH from the pituitary in 1955 (Saffran 1955), it was not until 1981 that CRF was isolated and
chemically characterized. Working with extracts derived from 500,000 sheep hypothalami, Vale
and colleagues at the Salk institute isolated, synthesized, and elucidated the structure of CRF
(Vale, et al. 1981). This discovery led to the availability of synthetic CRF, which allowed a
comprehensive assessment of the HPA axis to proceed. It is now clear CRF coordinates the
endocrine, immune, autonomic and behavioral responses of mammals to stress. The regulation
of CRF transcription is under control of a number of promoter elements. A cyclic AMP response
element (CRE) is located in the 5'flanking region of the human CRF gene, consistent with
evidence that protein kinase A (PKA) activity regulates CFR gene expression. A glucocorticoid
response element (GRE) is also located in the 5' flanking region of the CRF gene, which is
apparently the substrate where glucocorticoids act to inhibit CRF gene transcription (Hauger
Two CRF receptor subtypes, CRF1 and CRF2, with distinct anatomical localization and
receptor pharmacology have been identified (Chalmers et al., 1996; Lovenberg et al., 1995;
Grigoriadis et al., 1996; Chang et al., 1993; Chen et al., 1993) in rats and humans. Both
receptors are G-protein coupled receptors and are positively coupled to adenylyl cyclase via Gs.
In addition, a putative CRF3 receptor has recently been identified in catfish (Arai 2001). The
CRF1 receptor is predominant expressed in the pituitary, cerebellum, and neocortex in the rat
(Primus, et al. 1997). A growing body of evidence from animal studies has shown that the CRF1
receptors may specifically mediate some of the anxiogenic-like behaviors observed after
administration of CRF (Heinrichs, et al. 1997). The CRF2 receptor family is composed of two
primary splice variants, CRF2A and CRF2B. The CRF2A receptor is more prevalent in subcortical
regions, such as the ventromedial hypothalamus, lateral septum, and dorsal raphe nucleus,
whereas CRF2B is more abundantly expressed in the periphery. A structurally related member of
the CRF peptide family, urocortin, has also been identified in the mammalian brain. The
endogenous neuropeptide urocortin has equally high affinity for both the CRF1 and CRF2
receptor subtypes (Vaughan, et al. 1995), whereas CRF displays a higher affinity at CRF1
receptors than it does at CRF2 receptors. The newly discovered urocortin II shows high
selectivity for CRF2A receptors, though its anatomic localization does not correlate precisely with
the distribution of the CRF2A receptor (Reyes 2001). With the discovery of a new ligand and a
putative third receptor in the CRF family, much of the pharmacology and functional interactions
between these ligands and receptors remains to be discovered.
The effects of changes in glucocorticoid availability
A deficiency of endogenous glucocorticoids produces overt clinical symptoms including
weakness, fatigue, hypoglycemia, hyponatremia, hyperkalemia, fever, diarrhea, nausea and
shock. This condition, also known as Addison's disease, is most often caused by autoimmune
destruction of the adrenal cortex. However it is important to note that abrupt withdrawal from
exogenous corticosteroids or ACTH can also induce an Addisonian crisis, because the exogenous
administration of these compounds suppresses endogenous HPA axis activity. This is why
tapering of the dose of adrenal steroids is essential before discontinuation. Glucocorticoid
deficiency may also produce mild to severe depression, or less commonly, psychosis.
Excessive glucocorticoid secretion leads to a number of characteristic symptoms
including moon facies, plaethoric appearance, truncal obesity, purple abdominal striae,
hypertension, protein depletion and signs of glucose intolerance or overt diabetes mellitus.
Psychiatric symptoms, specifically depression and anxiety, are also associated with
glucocorticoid excess. Cognitive impairment, especially decrements in memory function and
attention are also common, and may be due to the direct effects of corticosteroids on the
hippocampal formation (Sadock 2000).
The most common form of non-iatrogenic hypercortisolism is due to an ACTH secreting
pituitary adenoma, also known as Cushing's disease. Harvey Cushing for whom the disease is
named, first documented the occurrences of psychiatric symptoms, particularly depression, in
1913 in his first description of the illness (1932). Other causes of hypercortisolism are often
referred to as Cushing's syndrome. Since Dr. Cushing's initial description, the occurrence of
depression in Cushing's syndrome has been well documented (Spillane, 1951; Zeiger et al.,
HPA axis abnormalities in depression
The occurrence of depression and other psychiatric symptoms in both Cushing's and
Addison's disease served as an impetus for researchers to scrutinize HPA axis abnormalities in
depression and other psychiatric disorders. Most investigators would agree that one of the most
venerable findings in all of psychiatry is the hyperactivity of the HPA axis observed in a
significant subset of patients with major depression [Table 2]. Based on the work of research
groups led by Board, Bunney and Hamburg, as well as by Carroll, Sachar, Stokes and Besser,
literally thousands of studies have been conducted in this area.
The earliest studies in this field demonstrated elevated plasma cortisol concentrations in
depressed patients (Carpenter and Bunney, 1971; Gibbons, 1962). Other markers of
hypercortisolism that have been reliably demonstrated in depressed patients include elevated 24-
hour urinary-free cortisol concentrations and increased levels of cortisol metabolites in urine
(Sachar, et al. 1970). One commonly used test to measure HPA axis function is the
dexamethasone suppression test (DST). In this test, 1 mg of dexamethasone is given at 11 P.M.,
blood is then drawn at 8 A.M. the following morning and cortisol levels measured.
Dexamethasone is a synthetic steroid similar to cortisol, and suppresses ACTH secretion, and
subsequently cortisol release, in healthy volunteers. Nonsuppression of plasma glucocorticoid
levels following the administration of dexamethasone is common in depression. The rate of
cortisol nonsuppression after dexamethasone administration generally correlates with the
severity of depression (Evans and Nemeroff 1987); in fact nearly all patients with major
depression with psychotic features exhibit DST non-suppresion (Arana, et al. 1985, Evans and
Nemeroff 1983). Since Carroll's initial report (Carroll, et al. 1968, Carroll, et al. 1968) and
subsequent claims for diagnostic utility (Carroll 1982), the dexamethasone suppression test has
generated considerable controversy (Arana and Mossman 1988) as to its diagnostic utility.
Diagnostic issues not withstanding, the overwhelming conclusion from the myriad of studies
demonstrates that a sizeable percentage of depressed patients exhibit HPA axis hyperactivity.
Further, the magnitude of the HPA axis hyperactivity also appears to correlate with the severity
of the depression.
Another method used to assess HPA axis activity is the CRF stimulation test, which
became available shortly after the synthesis of CRF. In this paradigm, CRF is administered
intravenously (usually a 1 μg/kg dose), and the ensuing ACTH and cortisol response is measured
at 30 minute intervals over a 2-3 hour period (Hermus, et al. 1984). Numerous studies have now
demonstrated a blunted ACTH and β-endorphin response to exogenously administered ovine
CRF (oCRF) or human CRF (hCRF) in depressed patients when compared to non-depressed
subjects, though the cortisol response in depressed patients and non-depressed control subjects
did not consistently differ (Amsterdam et al., 1988; Gold et al., 1984; Holsboer et al., 1984a;
Kathol et al., 1989; Young et al., 1990). The attenuated ACTH response to CRF is presumably
due to either chronic hypersecretion of CRF from nerve terminals in the median eminence, which
results in down-regulation of CRF receptors in the anterior pituitary, and/or to the chronic
hypercortisolemia. This receptor down-regulation results in a reduced responsivity of the
anterior pituitary to CRF, as has been demonstrated in laboratory animals (Aguilera, et al. 1986,
Holmes, et al. 1987, Wynn, et al. 1983, Wynn, et al. 1984, Wynn, et al. 1988). Following
recovery from depression, the documented disturbances in the HPA axis generally remit.
A combined dexamethasone/CRF test has also been developed. In this test, 1.5 mg of
dexamethasone is administered orally at night (23:00 h), and subjects receive an i.v. bolus of 100
μg of human CRF at 15:00 h the following day. Patients with HPA axis dysfunction, which is
frequently encountered in depression, display a paradoxically increased release of ACTH and
cortisol relative to controls. These abnormalities disappear following remission of depression,
and normalization of HPA axis function seems to precede full clinical remission (Holsboer 2000,
Heuser, et al. 1994). The combined DEX/CRF test appears to have much higher sensitivity for
detecting subtle alterations in HPA axis function; and approximately 80% of patients with major
depression exhibit an abnormal response to the DEX/CRF test. In contrast, only approximately
44% of patients with major depression demonstrate an abnormal response when the
dexamethasone suppression test is administered alone (Holsboer 2000, Heuser, et al. 1994).
Furthermore, otherwise healthy individuals with first degree relatives with an affective illness,
which greatly increases their own risk for psychiatric disorders, demonstrated cortisol and ACTH
responses to the DEX/CRF test which were higher than a control group, but less than patients
currently suffering from major depression. This suggests that a genetically transmittable defect
in corticosteroid receptor function may render these individuals more susceptible to developing
affective disorders (Holsboer, et al. 1995).
Structural changes in the components of the HPA axis have also been documented in
depressed patients. Perhaps in part due to the trophic effects of CRF, pituitary gland
enlargement has been documented in depressed patients as measured by MRI (Krishnan, et al.
1991). Enlargement of the adrenal glands, presumably due to ACTH hypersecretion, has
repeatedly been demonstrated in both depressed patients post mortem (Nemeroff, et al. 1992,
Amsterdam, et al. 1987) and in suicide victims (Dorovini-Zis and Zis 1987). It is reasonable to
hypothesize that the normal plasma cortisol response to CRF seen in depressed patients is due to
adrenocortical hypertrophy, in light of the blunted ACTH and β-endorphin responses to CRF
seen in these same patients (Gold, et al. 1984, Kathol, et al. 1989, Amsterdam, et al. 1987, Gold,
et al. 1986, Holsboer, et al. 1984). Presumably, although the ACTH response to CRF is
decreased in depressed patients, the enlarged adrenal cortex may secrete relatively greater
quantities of cortisol when compared to control subjects in response to a given amount of ACTH.
There are reports of increased cortisol responses to pharmacological doses of ACTH that support
this hypothesis (Amsterdam, et al. 1983, Jaeckle, et al. 1987, Kalin, et al. 1982, Krishnan, et al.
1990, Linkowski, et al. 1985), though discordant findings have also been reported (Heim, et al.
The studies thus far discussed focused primarily on dysregulations of the HPA axis, but
as mentioned earlier, CRF controls not only the neuroendocrine, but also the autonomic,
immune, and behavioral responses to stress in mammals. Moreover, results from both clinical
studies, and a rich body of literature conducted primarily in rodents and lower primates have
indicated the importance of CRF at extrahypothalamic sites. In rodents, primates, and humans,
CRF and its receptors have been heterogeneously localized in a variety of regions including the
amygdala, thalamus, hippocampus, and prefrontal cortex, among others (Suda et al., 1984;
Sanchez et al., 1999; Van Pett, 2000; Charlton et al., 1987). These brain regions are important
in regulating many aspects of the mammalian stress response, and in regulating affect. The
presence of CRF receptors in both the dorsal raphe (DR) and locus coeruleus (LC), the major
serotonergic and noradrenergic containing regions in the brain, respectively, also deserves
comment. Because most available antidepressants including the tricyclics and selective
serotonin reuptake inhibitors (SSRI) are believed to work via modulation of noradrenergic and/or
serotonergic systems, the neuroanatomical proximity of CRF and monoaminergic systems
suggests a possible site of interaction between CRF systems and antidepressants.
Involvement of extrahypothalamic CRF systems in the pathophysiology of depression is
suggested by numerous studies showing elevated CRF concentration in the cerebrospinal fluid
(CSF) of patients suffering from depression (Banki et al., 1987; Arato et al., 1989; France et al.,
1988; Nemeroff, 1988; Risch et al., 1992), though discrepant results have been reported (Roy, et
al. 1987). Elevated CSF CRF levels have also been detected in depressed people who committed
suicide (Arato, et al. 1989). A reduction in concentrations of CRF in CSF has been reported in
healthy volunteers treated with the tricyclic antidepressant desipramine (Veith, et al. 1993)
providing further evidence of a possible interconnection between antidepressants, noradrenergic
neurons and CRF systems. Similar effects have been reported with fluoxetine and ECT in
depressed patients (Nemeroff, et al. 1991).
Depressed patients who are nonsuppressors on the dexamethasone suppression test (DST)
also have significantly higher levels of CSF CRF than depressed patients with normal DST
results. Presumably the elevated CSF concentrations of CRF are due to CNS CRF
hypersecretion (Post, et al. 1982), which may be acting at sites throughout the brain and
contribute to many of the behaviors characteristic of depression. A reduction in the density of
CRF receptors in the frontal cortex has also been reported in the frontal cortex of suicide victims
(Nemeroff, et al. 1988). Presumably hypersecretion of CRF results in a downregulation of CRF
receptors in the frontal cortex.
While the exact mechanism contributing to CRF hyperactivity remains obscure, studies
from our group and others have document long-term persistent increases in HPA axis activity
and extrahypothalamic CRF neuronal activity after exposure to early untoward life events—for
example, neglect and child abuse respectively in both laboratory animals (rat and non-human
primates) and patients (Holsboer, et al. 1995, Nemeroff 1999, Holsboer, et al. 1987, Coplan, et
al. 1996). Early life stress apparently permanently sensitizes the HPA axis and leads to a greater
risk of developing depression later in life. In one schema, early sensitization of CRF systems
results in heightened responses to stress later in life. To measure HPA responsivity to stress, the
Trier Social Stress Test (TSST) was developed. This laboratory paradigm involves a simulated
10 minute public speech and a mental arithmetic task. The TSST has been validated as a potent
activator of the HPA axis in humans (Kirschbaum, et al. 1993). Recently, our group has reported
increased plasma adrenocorticotropin hormone (ACTH) and cortisol concentrations, presumably
due to hypersecretion of CRF, after exposure to the TSST in women (both depressed and non-
depressed) who were exposed to severe physical and emotional trauma as children (Heim, et al.
2000). These data provide evidence for functional hyperactivity of CRF systems that may be
influenced by early adverse life events.
Space constraints do not permit an extensive review of the preclinical literature, however
several additional points are worth interjecting. Numerous studies have documented that when
CRF is directly injected into the CNS of laboratory animals it produces effects reminiscent of the
cardinal symptoms of depression including decreased libido, reduced appetite and weight loss,
sleep disturbances, and neophobia. Indeed, newly developed CRF1 receptor antagonists
represent a novel putative class of antidepressants. Such compounds show activity in nearly
every preclinical screen for antidepressants and anxiolytics currently employed. Recently, a
small open label study examining the effectiveness of R121919, a CRF1 receptor antagonist, in
major depression was completed (Zobel, et al. 2000). Severity measures of both anxiety and
depression were reduced in the depressed patients. Although this drug is no longer in clinical
development, it is clear that CRF1 antagonists may represent a new class of psychotherapeutic
agents to treat anxiety and affective disorders.
HPA Axis alterations in other psychiatric disorders
Patients with other psychiatric disorders also exhibit HPA axis dysregulation, although
the vast majority of the data is concerned with HPA axis alterations in depression. When
depression is comorbid with a variety of other disorders such as multiple sclerosis, Alzheimer's
disease, multi-infarct dementia, Huntington's disease, and others, both CRF hypersecretion and
HPA axis hyperactivity are common. In contrast, HPA axis dysfunction has rarely been reported
in schizophrenia. Consistent with the role of CRF in both depression-like and anxiety-like
behaviors in preclinical animal studies, increased CSF CRF concentrations have been reported in
post-traumatic stress disorder (PTSD) (Bremner, et al. 1997). A recent elegant study using
indwelling cannula in the lumbar space, allowing repeated sampling of CSF several hours after
the initial, and presumably stressful, lumbar puncture, demonstrated elevated CSF CRF levels in
PTSD combat veterans (Baker, et al. 1999). In contrast, low serum cortisol and urinary free
cortisol levels have been repeatedly, yet unexpectedly, been detected in PTSD. One possible
mechanism that has been proposed by Yehuda and colleagues suggests heightened negative
feedback within the HPA axis in chronic PTSD patients (Yehuda, et al. 1996). Finally, CRF
neuronal degeneration is now well known to occur in the cerebral cortex of patients with
Alzheimer's disease with compensatory upregulation of CRF receptor numbers, and this effect
precedes the better-studied cholinergic neuronal involvement (Bissette 1998).
OVERVIEW OF HPT AXIS COMPONENTS AND FUNCTION
The thyroid gland, composed of two central lobes connected by an isthmus, synthesizes
the hormones thyroxine (T4) and triiodothyronine (T3). These iodine-containing compounds
serve as global regulators of the body's metabolic rate, and are also critical for brain
development. The release and synthesis of these hormones is ultimately controlled by signals
from the central nervous system.
The hypothalamic-pituitary-thyroid (HPT) axis is composed of three main parts, as its
name suggests. The tripeptide (pGlu-His-Pro-NH2) thyrotropin-releasing hormone (TRH) is
synthesized predominantly in the paraventricular nucleus in the hypothalamus and stored in
nerve terminals in the median eminence where it is released into the vessels of the hypothalamo-
hypophyseal portal system [Figure 3]. TRH is then transported to the sinusoids in the anterior
pituitary where it binds to thyrotropes and releases the peptide thyroid stimulating hormone
(TSH) into the systemic circulation. TRH is heterogeneously distributed in the brain which
strongly suggests a role for this peptide as a neurotransmitter, as well as a releasing hormone.
Thus TRH itself can produce direct effects on the CNS independent of its actions on pituitary
thyrotrophs. The HPT axis exhibits an ultradian rhythm where TSH secretion, and consequently
T3 and T4 levels, rise in the afternoon and evening, peak sometime after midnight and decline
throughout the day (Veldhuis 2000).
TSH is a 28 kDA glycoprotein composed of two noncovalently linked protein chains,
TSH-α and TSH-β. The α subunit is identical to the α subunit contained in other pituitary
hormones including follicle-stimulating hormone, luteinizing hormone and human chorionic
gonadotropin. Upon release from the pituitary, TSH circulates through the blood and exerts its
effects via binding to the TSH receptor in the thyroid, a G-protein coupled receptor that
stimulates the activation of adenylate cyclase.
Upon stimulation by TSH, the thyroid gland releases the iodinated amino acids T3 and T4.
Of the two hormones, T3 is much more physiologically active. Although debate still exists in the
literature, T4 is often considered a prohormone that becomes active after monodeiodination in
peripheral tissues. Thyroid hormones influence gene expression via two major thyroid hormone
receptors, TRα-1 and TRβ-1, which in turn bind to a specific DNA element, known as thyroid
response elements (TREs) located in the promoter regions of a diverse number of genes. These
receptors can function as homodimers, or bind with other nuclear factors such as thyroid
hormone receptor auxiliary proteins, as heterodimers to modulate the transcription of target
genes (Nestler 2001). T3 directly regulates the HPT axis by inhibiting TSH release and gene
expression in the pituitary, and TRH gene expression in the hypothalamus (DeVito 2000). This
is characteristic of the end-product negative feedback seen in the hypothalamic-pituitary-end
organ axes. In the circulation, these hormones are primarily bound to a carrier-protein,
thyroglobulin, though it is the unbound form of these hormones that are metabolically active.
Thyroid hormones have numerous effects on metabolism and increase heat production, oxygen
consumption, lipid metabolism, intestinal absorption of carbohydrates, cardiac function, and the
activity of the Na+-K+ ATPase. All of these functions are consistent with increasing metabolic
DISORDERS OF THE HPT AXIS
Disorders of the HPT axis lead to numerous psychiatric manifestations ranging from mild
depression to overt psychosis. Numerous conditions can lead to hypothyroid states, also known
as myxedema, including CNS causes of decreased TSH or TRH secretion, severe iodine
deficiency, thyroid surgery, drugs, or autoimmune disorders. The most common cause of
hypothyroidism is Hashimoto's thyroiditis, which is due to autoimminue destruction of thyroid
tissue. Regardless of the etiology, hypothyroidism leads to a number of clinical manifestations
including slowed mentation, forgetfulness, decreased hearing, cold intolerance, and ataxia.
Decreased energy, weight gain, depression, cognitive impairment or overt psychosis
("myxedemia madness") may also result. Due to the overlapping symptoms with clinical
depression, thyroid hormone deficiency must be ruled out when evaluating patients with
Hypothyroidism is frequently subclassified into the following 4 groups:
♦ Grade 1 hypothyroidism is classic primary hypothyroidism (increased TSH), decreased
peripheral thyroid hormone (T3 and T4) concentrations, and an increased TSH response to TRH
♦ Grade 2 hypothyroidism is characterized by normal, basal thyroid hormone concentrations,
but an increase in basal TSH concentrations and an exaggerated TSH response to TRH.
♦ Grade 3 hypothyroidism can only be detected by a TRH-stimulation test; basal thyroid
hormone and TSH concentrations are normal, but the TSH response to TRH is exaggerated
♦ Grade 4 hypothyroidism is defined as normal findings on the three thyroid axis function
tests noted above, but the patients have the abnormal presence of anti-thyroid antibodies.
Without treatment, most patients will progress from grade 4 to grade 1 hypothyroidism.
The first treatments for hypothyroidism became available in the 1890s; prior to that many
patients with this condition spent their final days in mental hospitals. One of the earliest
descriptions of the effects of treatment with thyroid extracts was reported by Shaw and Stansfield
in 1892. These physicians studied the effects of thyroid extracts in a patient suffering from
severe thyroid deficiency secondary to trauma to her thyroid gland. Within 10 weeks time
following treatment with a sheep thyroid extract, the mental signs associated with myxedema
disappeared in this patient and she was discharged (Shaw 1892). Stansfield followed this
patient's progress for several months, and five months after the last injection of thyroid extract,
symptoms of hypothyroidism began to recur. Following ingestion of additional thyroid extracts,
the symptoms were once again ameliorated. These results clearly demonstrated the profound
psychiatric effects of thyroid deficiency, and provided an early demonstration that treatment of
primary endocrine abnormalities can resolve the psychiatric manifestations of the disease
The first prospective study that scrutinized psychiatric comorbidity in patients with
hypothyroidism was carried out by Whybrow and colleagues (1969). In this seminal study, 5 of
the 7 patients manifested symptoms of depression at the time of the evaluation, while 6 of the 7
displayed cognitive impairment. Interestingly, of the 4 patients with depression who were
followed, thyroid replacement alone ameliorated the symptoms of depression in all of the
patients. In a later study, Jain et al (1972) studied 30 hypothyroid patients; in this study, 13 of 30
(43%) of the patients had a clinical depression, 10 (30%) had symptoms of anxiety, and 8 (27%)
were confused. Furthermore, these symptoms were improved or resolved following treatment of
the thyroid condition alone. These early studies clearly demonstrated that hypothyroid states
have pronounced psychiatric manifestations, predominantly depression and dementia, which can
be reversed following thyroid hormone replacement. Later studies have demonstrated varying
degrees of cognitive disturbance in up to 48% of psychiatrically ill hypothyroid cases (Boswell
2001), and approximately 50% of unselected hypothyroid patients have symptoms characteristic
of depression (Boswell 2001). Anxiety symptoms are also common, occurring in up to 30% of
unselected patients. Mania and hypomanic states have been rarely reported in hypothyroid
patients. Finally, although psychosis is the most common reported symptom in the case
literature on hypothyroidism (52.9%), it only accounts for approximately 5% of the psychiatric
morbidity in an unselected sample (Boswell 2001), presumably due to reporting bias.
HPT Axis dysfunction in patients with primary psychiatric disorders
Excluding patients with primary endocrine disorders, considerable data has revealed an
elevated rate of HPT axis dysfunction, predominantly hypothyroidism, in patients with major
depression [Table 3]. More than 25 years ago research groups led by Prange and Kastin
demonstrated that approximately 25% of patients with major depression exhibit a blunted TSH
response to TRH (Prange, et al. 1972, Kastin, et al. 1972). Presumably this is due to
hypersecretion of TRH from the median eminence which leads to TRH receptor downregulation
in the anterior pituitary resulting in reduced sensitivity of the pituitary to exogenous TRH. This
hypothesis seems plausible in light of evidence showing elevated TRH concentrations in the
cerebrospinal fluid (CSF) of drug-free depressed patients (Banki, et al. 1988). Depressed
patients have also been shown to have an increased occurrence of symptomless autoimmune
thyroiditis (SAT), defined by the abnormal presence of antithyroglobulin and/or antimicrosomal
thyroid antibodies consistent with Grade 4 hypothyroidism (Nemeroff, et al. 1985).
Recently, Duval et al (1996) performed a standard TSH stimulation test at both 8 A.M.
and 11 P.M. in a depressed patient population and normal controls. The difference between the
ΔTSH from 11 P.M. and the ΔTSH at the 8 A.M. time point was defined as ΔΔTSH. These
researchers demonstrated that depressed patients had a much lower ΔΔTSH than did controls.
Normal HPT axis function returned following remission from depression, but patients who did
not respond to antidepressant medications continued to show blunted ΔΔTSH. This suggests
that treatment with antidepressants per se is not responsible for the improvement in HPT axis
function. Further, patients with the lowest pretreatment evening thyrotropin secretion also had
the lowest rate of antidepressant response. This new methodology may serve as a more
sensitive method to detect changes in HPT axis function.
Interestingly, Post's group measured both cerebral blood flow and cerebral glucose
metabolism using positron emission tomography (PET) in both clinically depressed and bipolar
patients. Both measures of cerebral activity were inversely correlated with serum TSH levels,
and the authors suggested that HPT axis function contributes to primary and secondary mood
disorders (Marangell, et al. 1997). Also the current literature has clearly demonstrated elevated
TRH release in some depressed patients, but whether this is a causative factor in depression
remains unknown. This same group proposed that elevated TRH levels might instead be a
compensatory response to depression. In fact they reported that a lumbar intrathecal infusion of
500 μg of TRH into medication-free inpatients with depression produced a clinically robust, but
short-lived, improvement in mood and suicidality (Marangell, et al. 1997). Although this work
is preliminary, it does suggest the development of a systemically administered TRH receptor
agonist may represent a novel class of antidepressant agents.
Bipolar disorder and HPT abnormalities
HPT axis abnormalities have also been reported in bipolar disorders. Both elevated basal
plasma concentrations of TSH and an exaggerated TSH response to TRH have been
demonstrated (Haggerty, et al. 1987, Loosen and Prange 1982). There is also evidence that
bipolar patients with the rapid cycling subtype have a higher prevalence rate of hypothyroidism
(Grades I, II and III) than bipolar patients who do not (Bauer, et al. 1990, Cowdry, et al. 1983).
A blunted or absent evening surge of plasma TSH, a blunted TSH response to TRH, (Sack, et al.
1988, Souetre, et al. 1988), and the presence of antithyroid microsomal and/or antithyroglobulin
antibodies (Lazarus, et al. 1986, Myers, et al. 1985) have also been demonstrated in bipolar
Treatment of Hypothyroid states:
As noted above, thyroid hormone extracts from sheep or cattle were the first treatments
used that demonstrated efficacy in ameliorating the signs and symptoms of hypothyroidism.
Several synthetic derivatives were introduced in the 1960's which quickly replaced desiccated
thyroid tissue for the treatment of patients with thyroid disease. Among these are levothyroxine
(Levoxyl, Levothroid, Synthroid), synthetic forms of thyroxine (T4) and liothyronine (Cytomel),
and the synthetic levorotary isomer of triiodothyronine (T3). Moreover, in part due to the
seminal work carried out by Prange and collaborators in the United States in the 1960's, the use
of thyroid hormones in augmenting antidepressant response in depression was established.
HYPERTHYROID STATES:
Although a number of conditions including pituitary adenomas can lead to hyperthyroid
states, the most common non-iatrogenic cause of thyroid hormone excess is Graves' disease. In
Graves' disease, the body generates an autoantibody to the TSH receptor which directly
stimulates thyroid follicular cells to secrete excessive amounts of T3 and T4. In this state, the
normal negative feedback T3 and T4 exert on TRH and TSH release is disrupted. The clinical
manifestations of thyroid hormone excess are exaggerations of the normal physiologic effects of
T3 and T4; they include diaphoresis, heat intolerance, fatigue, dyspnea, palpitations, weakness
(especially in proximal muscles), weight loss despite an increased appetite, hyperdefecation,
increased psychomotor activity, and visual complaints. Psychiatric manifestations are also
common and include anxiety (13% of unselected cases), depression (28% of patients), and
cognitive changes (approximately 7% of patients). Psychotic manifestations and mania are less
common, occurring in only 2% of unselected cases. Overall psychiatric morbidity is much less
common in hyperthyroid states relative to hypothyroid states (Boswell 2001).
HPT Axis: Conclusions
Overall, there is clear evidence linking psychiatric symptomatology and thyroid disorders
that extends back over 100 years. The observations that hypothyroid patients exhibit symptoms
reminiscent of major depression led to a search for thyroid axis abnormalities in patients with
affective illness. The efficacy of thyroid augmentation in the treatment of depression (Dording
2000) and other affective disorders provides further evidence linking HPT axis function and
psychiatric illness (Prange 1996). Though work over the past 40 years has demonstrated a
number of HPT axis abnormalities in depressed and bipolar patients, the etiologic connection
between these findings remains elusive.
The overall organization of the HPG axis is similar to the other major neuroendocrine
axes. A "pulse" generator in the arcuate nucleus of the hypothalamus controls gonadotropin-
releasing hormone (GnRH) secretion, which occurs in a pulsatile fashion (Knobil 1990) in
intervals of 60-100 minutes (Nestler 2001). GnRH, previously known as luteinizing hormone-
releasing hormone (LHRH), is released into the portal circulation connecting the hypothalamus
and anterior pituitary where it binds to gonadotrophs and promotes the release of luteinizing
hormone (LH) and follicle-stimulation hormone (FSH) into the systemic circulation (Midgley
and Jaffe 1971). These hormones then bind to Leydig cells in the testes to promote testosterone
synthesis and secretion from Leydig cells and the ovaries to promote estrogen secretion. In
females, FSH also promotes the development of ovarian follicles and the synthesis and secretion
of androgen-binding proteins and inhibin. Inhibin acts directly on the anterior pituitary to inhibit
FSH secretion without affecting LH release. In both sexes, testosterone/estradiol generated by
the testes/ovaries feedback on the pituitary and hypothalamus to inhibit further FSH, LH and
GnRH release. Gonadal steroids, like glucocorticoids, modulate gene transcription. Gonadal
steroids can bind to androgen or estrogen response elements located in the regulatory regions of
specific genes and directly modulate the expression of those genes. Gonadal steroids may also
interact with transcription factors such as AP-1 or CREB, in turn influencing the expression of
the genes controlled by those transcription factors (Nestler 2001).
Despite the significantly higher rates of depression in women, data on HPG abnormalities
in psychiatric disorders remains remarkably limited. Early studies showed no differences in
plasma concentrations of LH and FSH in depressed postmenopausal women compared with
nondepressed matched control subjects (Nathan 1995). However a later studied showed
decreased plasma LH concentrations in depressed postmenopausal women compared to matched
controls (Brambilla, et al. 1990). In a more recent study, significantly lower estradiol levels
were detected in women with depression, but the blood levels of other reproductive hormones
fell within the normal range (Young, et al. 2000). Because estradiol affects a number of
neurotransmitter systems including norepinephrine and serotonin, these results merit further
The response to exogenous administration of GnRH in depressed patients has also been
investigated. Normal LH and FSH responses to a high dose of GnRH (i.e. 250 μg) have been
reported in male depressed and female depressed (pre and post menopausal) patients (Winokur,
et al. 1982), whereas a decreased LH response to a lower dose of GnRH (150 μg) has been
reported in pre- and postmenopausal depressed patients (Brambilla, et al. 1990). Unden and
colleagues observed no change in basal or TRH/LHRH stimulated LH concentrations in a
depressed cohort including both sexes, though depressed males with an abnormal dexamethasone
suppression test response showed a significantly higher increase in FSH compared to the controls
(Unden, et al. 1988).
The prevalence of mood disorders in women including premenstrual syndrome and
postpartum depression also deserves mention. Premenstrual dysphoric disorder (PMDD) is a
cyclic recurrence of symptoms, which are both somatic (edema, fatigue, breast tenderness,
headaches) and psychological (depression, irritability, and affective liability). The symptoms
start following ovulation and disappear within the first day or two of menses followed by a
symptom-free interval between menses and the next ovulation. In some cases (5 – 10%)
symptoms may be severe enough to interfere with normal functioning leading to the diagnosis of
PMDD (Altshuler, et al. 1995). GnRH agonists that produce a "clinical ovariectomy" by
downregulation of GnRH receptors in the pituitary and reduced gonadotropin secretions have
been shown to be an effective treatment for premenstrual syndrome (PMS) suggesting that the
HPG axis is involved in the manifestation of symptoms (Freeman, et al. 1997). However
significant variations in HPG axis function have yet to be identified in women especially
susceptible to PMS.
Postpartum mood disorders are also common, occurring in approximately 10% of women
after childbirth. Both postpartum depression and the less frequent postpartum psychosis occur
with highest prevalence in the first three months after childbirth (Wisner and Stowe 1997). The
timing of these syndromes would suggest that neuroendocrine dysregulation may contribute to
the expression of such disorders, but no major abnormalities in HPG axis function were detected
in a prospective investigation of postpartum disorders (O'Hara, et al. 1990). Additional research
on the HPG axis in depression and in other mood states is needed.
Hypothalamic-Prolactin Axis
Unlike other anterior pituitary hormones, prolactin release is regulated via tonic
inhibition by prolactin-inhibitory factor (PIF), which was later determined to be dopamine.
Dopamine neurons in the tuberoinfundibular system of the hypothalamus directly inhibit
prolactin release. Prolactin can also inhibit its own release by a short-loop negative feedback to
the hypothalamus. Prolactin primarily regulates the behavioral aspects of reproduction and
infant care. Serum prolactin levels are normally low through life in males. Basal prolactin levels
increase in females following parturition, and suckling stimulates prolactin release. Prolactin
itself stimulates breast growth and milk synthesis. TRH, oxytocin, serotonin, estrogen and other
neuroregulators have prolactin-releasing factor activity (Fink 2000).
Excess circulating prolactin can lead to a number of clinical symptoms. The most
common causes of hyperprolactinemia are tumors, usually microadenomas of pituitary
lactotrophs, or following treatment with conventional antipsychotic medications because of their
potent blockade of dopamine receptors. Hyperprolactinemia often leads to reduced testosterone
secretion in men, and a decreased libido in both men and women. Patients may also complain of
depression, stress intolerance, anxiety and increased irritability that usually resolve following
treatments that reduce serum prolactin levels. Despite these effects, alterations in the
hypothalamic-prolactin axis have not been clearly demonstrated in psychiatric disorders
(Nicholas, et al. 1998). Because prolactin release is inhibited by dopamine, the prolactin
response to infusions of dopaminergic agonists has also been used to estimate CNS
dopaminergic tone, though it likely only reflects hypothalamic dopamine neuronal function.
Although abnormalities in prolactin secretion have not been clearly demonstrated in
depression per se, a large number of reports have used provocative tests of prolactin secretion in
patients with psychiatric disorders (For a review see (Van de Kar 1989)). Briefly, these tests use
agents that increase serotonergic transmission, for example L-trypotophan, 5-hydroxytryptophan
(5-HTP), and fenfluramine, among others. In general the prolactin response to agents that
increase serotonergic activity is blunted in depression (Mann et al., 1995; Golden et al., 1992), as
well as in patients with cluster-B personality disorders (Coccaro, et al. 1997). This data suggests
that the blunted prolactin response is mediated by alteration in 5-HT1A receptor responsiveness
and that serotonergic transmission in these patients is dysfunctional.
OXYTOCIN AND VASOPRESSIN
Oxytocin and arginine-vasopressin (AVP), also known as anti-diuretic hormone (ADH),
are nona-peptides synthesized in the magnocellular neurons of the paraventricular nucleus of the
hypothalamus, and released directly into the bloodstream from axon terminals in the posterior
pituitary. This is in contrast to the hypothalamic releasing factors we have discussed thus far
which are released in the portal system from the median eminence and distinct from the anterior
pituitary hormones that are released following the activation of pituicytes by the releasing factors
synthesized in the hypothalamus.
AVP has prominent roles in controlling fluid balance via its effects on the kidney, in
regulating blood pressure by its vasoconstrictive effects on blood vessels, and can directly
promote the sensation of thirst. AVP also promotes the release of ACTH from the anterior
pituitary in the presence of CRF, and is released following stressful stimuli (Insel 1997). In
humans, oxytocin is predominantly involved in controlling smooth muscle contraction during
parturition (myometrium), and during both breast-feeding, by mediating milk letdown in
lactating mothers. In rodents, oxytocin promotes a number of reproductive (grooming, arousal,
lordosis, orgasm, nesting, birthing) and maternal behaviors.
Although there are marked species differences in the effects of oxytocin, central infusion
of this peptide in females of a monogamous prairie vole species promotes life-long pair bonding
in the absence of mating. Furthermore, pair bonding in this species, which normally
accompanies mating, can be blocked by oxytocin antagonists thus implicating oxytocin's key
role in the expression of this lifelong behavior. Rodent studies have also demonstrated that AVP
has a pair-bonding function in males, analogous to the pair-bond promoting behaviors induced
by oxytocin administration in females. AVP promotes monogamy and paternal behavior in male
prairie voles. These studies have led some researchers to speculate that oxytocin and AVP may
play a role in psychiatric disorders characterized by disrupted affiliative behaviors such as
Asperger's disease and autism (Insel 1997). Clearly, more work is needed in order to better
understand the function of these two hormones in the human brain.
The Pituitary Growth Hormone Axis
Growth hormone (GH) is synthesized and secreted from somatotrophs located in the
anterior pituitary. Its release is unique in that it is controlled by two peptide hypothalamic
hypophysiotropic hormones, growth-hormone-releasing factor (GHRF) and somatostatin.
Somatostatin, also known as growth hormone-release-inhibiting hormone (GHIH) or
somatotropin release-inhibitory factor (SRIF), was first isolated from ovine hypothalamus in
1974. It is a tetradecapeptide, containing a disulfide bridge linking the two cysteine residues.
Somatostatin is released predominantly from the periventricular and paraventricular nucleus of
the hypothalamus and inhibits GH release. Somatostatin has a wide extrahypothalamic
distribution in brain regions including the cerebral cortex, hippocampus, and amygdala.
GHRF, also known as growth hormone releasing hormone (GHRH), was characterized
and sequenced in 1981 after considerable difficulty. The long-postulated GHRF was
discovered several years after the elucidation of the structure of somatostatin, from extracts of an
ectopic tumor associated with acromegaly. GHRF is a 44 amino acid peptide, and has the most
limited CNS distribution of all the hypothalamic-releasing hormones that have been identified.
GHRF containing neurons are concentrated in the infundibular and arcuate nuclei of the
hypothalamus and stimulate the synthesis and release of GH. Dopamine, norepinephrine and
serotonin innervate GHRF-containing neurons to modulate GH release. Both GHRF and SRIF
are released from the median eminence into the hypothalamo-hypophyseal portal system where
they act on somatotrophs in the anterior pituitary to regulate GH release. Negative feedback is
provided by GH, which stimulates somatostatin release preventing further GH release. The
growth hormone axis is unique in that it does not have a single target endocrine gland but instead
growth hormone acts directly on targets including bone, muscle and liver. GH also stimulates
the release of somatomedin from the liver and insulin like growth factors.
Growth hormone is released in a pulsatile fashion, with highest release occurring around
the time of sleep onset and extending into the first non-REM period of sleep (Finkelstein, et al.
1972). A variety of stressors including starvation, exertion, or emotional stress also promote
growth hormone release (Nestler 2001). Growth hormone is necessary for the longitudinal bone
growth which occurs during late childhood; accordingly GH levels are high in children, reach
their peak during adolescence and decline throughout adulthood. In additions to its effects on the
long bones, growth hormone has predominantly anabolic effects and leads to increased muscle
mass and decreased body fat.
Growth hormone release to a variety of stimuli including L-Dopa, a DA precursor (Boyd,
et al. 1970), apomorphine, a centrally active DA agonist (Fink 2000), and the serotonin
precursors L-tryptophan (Muller, et al. 1974) and 5-hydroxytryptophan (Imura, et al. 1973) has
been demonstrated. Several findings indicate dysregulation of growth hormone secretion in
depression [Table 4]. Studies have demonstrated a blunted nocturnal GH surge in
depression(Schilkrut, et al. 1975), whereas daylight GH secretion seems to be exaggerated in
both unipolar and bipolar depressed patients (Mendlewicz, et al. 1985). A number of studies
have also demonstrated a blunted GH response to the α-adrenergic agonist clonidine in depressed
patients (Siever, et al. 1982, Charney, et al. 1982). Siever et al. (1982) demonstrated the blunted
GH response to clonidine was not related to age or sex, and this study provided evidence that the
diminished GH response to clonidine may be secondary to decreased α2-adrenergic receptor
sensitivity in depression. Using a GHRF stimulation test, our group later demonstrated a slight
exaggeration of GH response to GHRF in depressed patients compared to controls, although this
group difference was mainly attributable to 3 of the 19 depressed patients who exhibited
markedly high GH responses to GHRF (Krishnan, et al. 1988). Others, however, have reported a
blunted GH response to GHRH in depressed patients. Thus it is unclear whether the blunted GH
response to clonidine seen in depression is due to a pituitary defect in GH secretion, further
implicating a subsensitivity of α-adrenergic receptors in depression, or to a GHRH deficit.
Recently a diminished GH response to clonidine was demonstrated in children and adolescents at
high risk for major depressive disorder. When considered with evidence demonstrating GH
dysregulation in childhood depression (Ryan, et al. 1994), it suggests the blunted GH response
seen in high-risk adolescents may represent a trait marker for depression in children and
adolescents (Birmaher, et al. 2000). Arguably, the blunted GH response to clonidine seen in
depression may be the most reproducible and specific finding in the biology of affective
A GHRH stimulation test has also been developed and studied in depressed patients.
Two groups have shown a blunted GH response to GHRH in depressed patients (Lesch, et al.
1987a, Lesch, et al. 1987b, Risch 1991). However Krishnan and colleagues (Krishnan, et al.
1988, Krishnan, et al. 1988) found minimal differences in serum GH response to GHRH in
between depressed and control patients. A comprehensive review of GHRH stimulation tests in
depression, anorexia nervosa, bulimia, panic disorder, schizophrenia, and Alzheimer's disease
was conducted and the authors concluded that the results of this test are not always consistent
and in some cases contradictory (Skare, et al. 1994). Factors including the variability of GHRH-
stimulated GH among controls, lack of standard outcome measures, and age and gender related
effects may account for some of this variability. Further studies using GHRH will help develop
a standard stimulation test to further clarify the response to GHRH in depression and other
psychiatric disorders.
Several studies have demonstrated decreased SRIF levels in the CSF of patients suffering
depression (Agren and Lundqvist 1984, Gerner and Yamada 1982), dementia, schizophrenia
(Bissette, et al. 1986) and Alzheimer's disease (Molchan, et al. 1993) (Bissette 1998).
Somatostatin concentrations are also markedly elevated in the basal ganglia of patients with
Huntington's disease (Nemeroff, et al. 1983), though the implications of this finding are
unknown. Somatostatin also inhibits the release of both CRF and ACTH (Brown, et al. 1984,
Heisler, et al. 1982, Richardson and Schonbrunn 1981) indicating a direct interaction between
the growth-hormone and HPA axes. No published studies measuring GHRH concentration and
GHRH-mRNA expression have been conducted in postmortem tissue of depressed patients and
matched controls which, in light of the evidence presented here, is of interest. Similarly, CSF
studies of GHRH are also lacking.
Summary and Conclusion
Basic clinical observations of psychiatric disorders associated with primary endocrine
disorders such as Cushing's syndrome and hypothyroidism has led to our broader understanding
of the role of neuroendocrine disturbances in a variety of psychiatric disorders including
depression and bipolar disorders. These studies have led to major advances in biological
psychiatry by helping to understand the brain circuits involved in the pathophysiology of mood
and anxiety disorders. Foremost among these is the CRF theory of depression, which is
supported by studies from a variety of disciplines, and which has led to the development of a
novel therapeutic approach, namely CRF receptor antagonists. Further this work has provided a
mechanism to explain the increase in depression seen in patients exposed to trauma early in life
(first postulated by Freud in the early part of the twentieth century). If CRF truly is the ‘black
bile' of depression, CRF antagonists may represent a novel class of antidepressants with a unique
mechanism of action from other commonly used antidepressants. Indeed, a number of CRF-
receptor antagonists are now in clinical development as novel anxiolytics and antidepressants.
In addition to the HPA axis and CRF alterations observed in depression, HPT axis
abnormalities are also very common; the majority of depressed patients, in fact, exhibit
alterations in one of these two axes. Furthermore, there is widely replicated blunting of growth-
hormone response to clonidine and the blunted prolactin response to serotonergic stimuli in
depressed patients. Although these studies have not added much understanding to the prevailing
monoamine theory of depression, the mechanistic studies that have followed have been
remarkably fruitful. Is it obvious that the vast majority of studies have been focused on patients
with mood disorders, particularly unipolar depression. Clearly other disorders including eating
disorders, anxiety disorders, schizophrenia, and axis II diagnoses should also be evaluated with
similar scrutiny.
The availability of selective ligands that can be utilized with positron-emission
tomography will mark the next major leap in our understanding of the neuroendocrine axes in
psychiatric disorders. The ability to determine peptide-receptor alterations in the brain and
pituitary of patients with psychiatric disorders will contribute immensely to our understanding of
the neurobiological underpinnings of such disorders.
Finally, a growing number of studies have demonstrated that depression is a systemic
disease that increases vulnerability to other disorders. Depressed patients demonstrate increased
incidence of coronary artery disease and stroke, osteoporosis, and perhaps cancer. These
observations may at least be partly attributed to the endocrine alterations observed in depression.
Acknowledgements:
We would like to thank Tomo Narashima and Julia Knox for their artistic assistance, and
acknowledge support from MH-42088 and the Conte Center for the Neuroscience of Mental
Disorders (MH-58922).
Bibliography:
Agren H, Lundqvist G (1984) Low levels of somatostatin in human CSF mark depressive
episodes. Psychoneuroendocrinology 9:233-248.
Aguilera G, Wynn PC, Harwood JP, Hauger RL, Millan MA, Grewe C, Catt KJ (1986)
Receptor-mediated actions of corticotropin-releasing factor in pituitary gland and nervous
system. Neuroendocrinology 43:79-88.
Altshuler LL, Hendrick V, Parry B (1995) Pharmacological management of premenstrual
disorder. Harvard Review of Psychiatry 2:233-245.
Amsterdam JD, Maislin G, Winokur A, Berwish N, Kling M, Gold P (1988) The oCRH
stimulation test before and after clinical recovery from depression. Journal of Affective
Disorders 14:213-222.
Amsterdam JD, Marinelli DL, Arger P, Winokur A (1987) Assessment of adrenal gland volume
by computed tomography in depressed patients and healthy volunteers: a pilot study.
Psychiatry Research 21:189-197.
Amsterdam JD, Winokur A, Abelman E, Lucki I, Rickels K (1983) Cosyntropin (ACTH alpha 1-
24) stimulation test in depressed patients and healthy subjects. American Journal of
Psychiatry 140:907-909.
Arana GW, Baldessarini RJ, Ornsteen M (1985) The dexamethasone suppression test for
diagnosis and prognosis in psychiatry. Commentary and review. Archives of General
Psychiatry 42:1193-1204.
Arana GW, Mossman D (1988) The dexamethasone suppression test and depression. Approaches
to the use of a laboratory test in psychiatry. Neurologic Clinics 6:21-39.
Arato M, Banki CM, Bissette G, Nemeroff CB (1989) Elevated CSF CRF in suicide victims.
Biological Psychiatry 25:355-359.
Baker DG, West SA, Nicholson WE, Ekhator NN, Kasckow JW, Hill KK, Bruce AB, Orth DN,
Geracioti TD, Jr. (1999) Serial CSF corticotropin-releasing hormone levels and
adrenocortical activity in combat veterans with posttraumatic stress disorder [published
erratum appears in Am J Psychiatry 1999 Jun;156(6):986]. American Journal of
Psychiatry 156:585-588.
Banki CM, Bissette G, Arato M, Nemeroff CB (1988) Elevation of immunoreactive CSF TRH in
depressed patients. American Journal of Psychiatry 145:1526-1531.
Banki CM, Bissette G, Arato M, O'Connor L, Nemeroff CB (1987) CSF corticotropin-releasing
factor-like immunoreactivity in depression and schizophrenia. American Journal of
Psychiatry 144:873-877.
Bauer MS, Whybrow PC, Winokur A (1990) Rapid cycling bipolar affective disorder. I.
Association with grade I hypothyroidism. Archives of General Psychiatry 47:427-432.
Birmaher B, Dahl RE, Williamson DE, Perel JM, Brent DA, Axelson DA, Kaufman J, Dorn LD,
Stull S, Rao U, Ryan ND (2000) Growth hormone secretion in children and adolescents
at high risk for major depressive disorder. Archives of General Psychiatry 57:867-872.
Bissette G, Cook, L., Smith, W., Dole, K.C., Crain, B., Nemeroff, C.B. (1998) Regional
neuropeptide pathology in Alzheimer's disease: corticotropin-releasing factor and
somatostatin. Journal of Alzheimer's Disease 1:1-15.
Bissette G, Widerlov E, Walleus H, Karlsson I, Eklund K, Forsman A, Nemeroff CB (1986)
Alterations in cerebrospinal fluid concentrations of somatostatinlike immunoreactivity in
neuropsychiatric disorders. Archives of General Psychiatry 43:1148-1151.
Boswell E, Anfinson, TJ., Nemeroff, CB (2001) Neuropsychiaric aspects of endocrine disorders.
In: Textbook of Neuropsychiatry, 3rd edition (Yudofsky S, Hales, R., ed). Washington,
DC: American Psychiatric Association Press, Inc.
Boyd AEd, Lebovitz HE, Pfeiffer JB (1970) Stimulation of human-growth-hormone secretion by
L-dopa. New England Journal of Medicine 283:1425-1429.
Brambilla F, Maggioni M, Ferrari E, Scarone S, Catalano M (1990) Tonic and dynamic
gonadotropin secretion in depressive and normothymic phases of affective disorders.
Psychiatry Research 32:229-239.
Bremner JD, Licinio J, Darnell A, Krystal JH, Owens MJ, Southwick SM, Nemeroff CB,
Charney DS (1997) Elevated CSF corticotropin-releasing factor concentrations in
posttraumatic stress disorder. American Journal of Psychiatry 154:624-629.
Brown MR, Rivier C, Vale W (1984) Central nervous system regulation of adrenocorticotropin
secretion: role of somatostatins. Endocrinology 114:1546-1549.
Carpenter WT, Jr., Bunney WE, Jr. (1971) Adrenal cortical activity in depressive illness.
American Journal of Psychiatry 128:31-40.
Carroll BJ (1982) Use of the dexamethasone suppression test in depression. Journal of Clinical
Psychiatry 43:44-50.
Carroll BJ, Martin FI, Davies B (1968a) Pituitary-adrenal function in depression. Lancet 1:1373-
Carroll BJ, Martin FI, Davies B (1968b) Resistance to suppression by dexamethasone of plasma
11-O.H.C.S. levels in severe depressive illness. British Medical Journal 3:285-287.
Chalmers DT, Lovenberg TW, Grigoriadis DE, Behan DP, De Souza EB (1996) Corticotrophin-
releasing factor receptors: from molecular biology to drug design. Trends in
Pharmacological Sciences 17:166-172.
Chang CP, Pearse RVd, O'Connell S, Rosenfeld MG (1993) Identification of a seven
transmembrane helix receptor for corticotropin-releasing factor and sauvagine in
mammalian brain. Neuron 11:1187-1195.
Charlton BG, Ferrier IN, Perry RH (1987) Distribution of corticotropin-releasing factor-like
immunoreactivity in human brain. Neuropeptides 10:329-334.
Charney DS, Heninger GR, Sternberg DE, Hafstad KM, Giddings S, Landis DH (1982)
Adrenergic receptor sensitivity in depression. Effects of clonidine in depressed patients
and healthy subjects. Archives of General Psychiatry 39:290-294.
Chen R, Lewis KA, Perrin MH, Vale WW (1993) Expression cloning of a human corticotropin-
releasing-factor receptor. Proceedings of the National Academy of Sciences of the United
States of America 90:8967-8971.
Coccaro EF, Kavoussi RJ, Hauger RL (1997) Serotonin function and antiaggressive response to
fluoxetine: a pilot study. Biological Psychiatry 42:546-552.
Coplan JD, Andrews MW, Rosenblum LA, Owens MJ, Friedman S, Gorman JM, Nemeroff CB
(1996) Persistent elevations of cerebrospinal fluid concentrations of corticotropin-
releasing factor in adult nonhuman primates exposed to early-life stressors: implications
for the pathophysiology of mood and anxiety disorders. Proceedings of the National
Academy of Sciences of the United States of America 93:1619-1623.
Cowdry RW, Wehr TA, Zis AP, Goodwin FK (1983) Thyroid abnormalities associated with
rapid-cycling bipolar illness. Archives of General Psychiatry 40:414-420.
DeVito W (2000) Neuroendocrine regulation of Thyroid Function. In: Neuroendocrinology in
Physiology and Medicine (Conn P, Freeman, ME, ed), pp 225-241. Totowa, New Jersey:
Dording CM (2000) Antidepressant augmentation and combinations. Psychiatric Clinics of
North America 23:743-755.
Dorovini-Zis K, Zis AP (1987) Increased adrenal weight in victims of violent suicide. American
Journal of Psychiatry 144:1214-1215.
Duval F, Mokrani MC, Crocq MA, Jautz M, Bailey P, Diep TS, Macher JP (1996) Effect of
antidepressant medication on morning and evening thyroid function tests during a major
depressive episode. Archives of General Psychiatry 53:833-840.
Evans DL, Nemeroff CB (1983) Use of the dexamethasone suppression test using DSM-III
criteria on an inpatient psychiatric unit. Biological Psychiatry 18:505-511.
Evans DL, Nemeroff CB (1987) The clinical use of the dexamethasone suppression test in DSM-
III affective disorders: correlation with the severe depressive subtypes of melancholia and
psychosis. Journal of Psychiatric Research 21:185-194.
Fink G (2000) Neuroendocrine Regulation of Pituitary Function: General Principles. In:
Neuroendocrinology in Physiology and Medicine (Conn P, Freeman, ME, ed), pp 112-
120. Totowa, NJ: Humana Press.
Finkelstein JW, Roffwarg HP, Boyar RM, Kream J, Hellman L (1972) Age-related change in the
twenty-four-hour spontaneous secretion of growth hormone. Journal of Clinical
Endocrinology & Metabolism 35:665-670.
France RD, Urban B, Krishnan KR, Bissett G, Banki CM, Nemeroff C, Speilman FJ (1988) CSF
corticotropin-releasing factor-like immunoactivity in chronic pain patients with and
without major depression. Biological Psychiatry 23:86-88.
Freeman EW, Sondheimer SJ, Rickels K (1997) Gonadotropin-releasing hormone agonist in the
treatment of premenstrual symptoms with and without ongoing dysphoria: a controlled
study. Psychopharmacology Bulletin 33:303-309.
Gerner RH, Yamada T (1982) Altered neuropeptide concentrations in cerebrospinal fluid of
psychiatric patients. Brain Research 238:298-302.
Gibbons J, McHugh, PR (1962) Plasma cortisol in depressive illness. Journal of Psychiatric
Research 1:162-171.
Gold PW, Chrousos G, Kellner C, Post R, Roy A, Augerinos P, Schulte H, Oldfield E, Loriaux
DL (1984) Psychiatric implications of basic and clinical studies with corticotropin-
releasing factor. American Journal of Psychiatry 141:619-627.
Gold PW, Loriaux DL, Roy A, Kling MA, Calabrese JR, Kellner CH, Nieman LK, Post RM,
Pickar D, Gallucci W (1986) Responses to corticotropin-releasing hormone in the
hypercortisolism of depression and Cushing's disease. Pathophysiologic and diagnostic
implications. New England Journal of Medicine 314:1329-1335.
Golden RN, Ekstrom D, Brown TM, Ruegg R, Evans DL, Haggerty JJ, Jr., Garbutt JC, Pedersen
CA, Mason GA, Browne J (1992) Neuroendocrine effects of intravenous clomipramine in
depressed patients and healthy subjects. American Journal of Psychiatry 149:1168-1175.
Grigoriadis DE, Lovenberg TW, Chalmers DT, Liaw C, De Souze EB (1996) Characterization of
corticotropin-releasing factor receptor subtypes. Annals of the New York Academy of
Sciences 780:60-80.
Haggerty JJ, Jr., Simon JS, Evans DL, Nemeroff CB (1987) Relationship of serum TSH
concentration and antithyroid antibodies to diagnosis and DST response in psychiatric
inpatients. American Journal of Psychiatry 144:1491-1493.
Hauger R, Dautzenberg, FM (2000) Regulation of the Stress Response by Corticotropin
Releasing Factor. In: Neuroendocrinology in Physiology and Medicine (Conn P,
Freeman, ME., ed), pp 267-293. Totowa, New Jersey: Humana Press.
Heim C, Newport DJ, Bonsall R, Miller AH, Nemeroff CB (2001) Altered pituitary-adrenal axis
responses to provocative challenge tests in adult survivors of childhood abuse. American
Journal of Psychiatry 158:575-581.
Heim C, Newport DJ, Heit S, Graham YP, Wilcox M, Bonsall R, Miller AH, Nemeroff CB
(2000) Pituitary-adrenal and autonomic responses to stress in women after sexual and
physical abuse in childhood. JAMA 284:592-597.
Heinrichs SC, Lapsansky J, Lovenberg TW, De Souza EB, Chalmers DT (1997) Corticotropin-
releasing factor CRF1, but not CRF2, receptors mediate anxiogenic-like behavior.
Regulatory Peptides 71:15-21.
Heisler S, Reisine TD, Hook VY, Axelrod J (1982) Somatostatin inhibits multireceptor
stimulation of cyclic AMP formation and corticotropin secretion in mouse pituitary tumor
cells. Proceedings of the National Academy of Sciences of the United States of America
Hermus AR, Pieters GF, Smals AG, Benraad TJ, Kloppenborg PW (1984) Plasma
adrenocorticotropin, cortisol, and aldosterone responses to corticotropin-releasing factor:
modulatory effect of basal cortisol levels. Journal of Clinical Endocrinology &
Metabolism 58:187-191.
Heuser I, Yassouridis A, Holsboer F (1994) The combined dexamethasone/CRH test: a refined
laboratory test for psychiatric disorders. Journal of Psychiatric Research 28:341-356.
Holmes MC, Catt KJ, Aguilera G (1987) Involvement of vasopressin in the down-regulation of
pituitary corticotropin-releasing factor receptors after adrenalectomy. Endocrinology
Holsboer F (2000) The corticosteroid receptor hypothesis of depression.
Neuropsychopharmacology 23:477-501.
Holsboer F, Lauer CJ, Schreiber W, Krieg JC (1995) Altered hypothalamic-pituitary-
adrenocortical regulation in healthy subjects at high familial risk for affective disorders.
Neuroendocrinology 62:340-347.
Holsboer F, Muller OA, Doerr HG, Sippell WG, Stalla GK, Gerken A, Steiger A, Boll E,
Benkert O (1984a) ACTH and multisteroid responses to corticotropin-releasing factor in
depressive illness: relationship to multisteroid responses after ACTH stimulation and
dexamethasone suppression. Psychoneuroendocrinology 9:147-160.
Holsboer F, Von Bardeleben U, Gerken A, Stalla GK, Muller OA (1984b) Blunted corticotropin
and normal cortisol response to human corticotropin-releasing factor in depression
[letter]. New England Journal of Medicine 311:1127.
Holsboer F, von Bardeleben U, Wiedemann K, Muller OA, Stalla GK (1987) Serial assessment
of corticotropin-releasing hormone response after dexamethasone in depression.
Implications for pathophysiology of DST nonsuppression. Biological Psychiatry 22:228-
Imura H, Nakai Y, Yoshimi T (1973) Effect of 5-hydroxytryptophan (5-HTP) on growth
hormone and ACTH release in man. Journal of Clinical Endocrinology & Metabolism
Insel TR (1997) A neurobiological basis of social attachment. American Journal of Psychiatry
Jaeckle RS, Kathol RG, Lopez JF, Meller WH, Krummel SJ (1987) Enhanced adrenal sensitivity
to exogenous cosyntropin (ACTH alpha 1-24) stimulation in major depression.
Relationship to dexamethasone suppression test results. Archives of General Psychiatry
Jain VK (1972) A psychiatric study of hypothyroidism. Psychiatria Clinica 5:121-130.
Kalin NH, Weiler SJ, Shelton SE (1982) Plasma ACTH and cortisol concentrations before and
after dexamethasone. Psychiatry Research 7:87-92.
Kastin AJ, Ehrensing RH, Schalch DS, Anderson MS (1972) Improvement in mental depression
with decreased thyrotropin response after administration of thyrotropin-releasing
hormone. Lancet 2:740-742.
Kathol RG, Jaeckle RS, Lopez JF, Meller WH (1989) Consistent reduction of ACTH responses
to stimulation with CRH, vasopressin and hypoglycaemia in patients with major
depression. British Journal of Psychiatry 155:468-478.
Kirschbaum C, Pirke KM, Hellhammer DH (1993) The 'Trier Social Stress Test'--a tool for
investigating psychobiological stress responses in a laboratory setting.
Neuropsychobiology 28:76-81.
Knobil E (1990) The GnRH pulse generator. American Journal of Obstetrics & Gynecology
Krishnan KR, Doraiswamy PM, Lurie SN, Figiel GS, Husain MM, Boyko OB, Ellinwood EH,
Jr., Nemeroff CB (1991) Pituitary size in depression [see comments]. Journal of Clinical
Endocrinology & Metabolism 72:256-259.
Krishnan KR, Manepalli AN, Ritchie JC, Rayasam K, Melville ML, Daughtry G, Thorner MO,
Rivier JE, Vale WW, Nemeroff CB (1988a) Growth hormone-releasing factor stimulation
test in depression. American Journal of Psychiatry 145:90-92.
Krishnan KR, Manepalli AN, Ritchie JC, Rayasam K, Melville ML, Thorner MO, Rivier JE,
Vale WW, Nemeroff CB (1988b) Growth hormone response to growth hormone-
releasing factor in depression. Peptides 9 Suppl 1:113-116.
Krishnan KR, Ritchie JC, Saunders WB, Nemeroff CB, Carroll BJ (1990) Adrenocortical
sensitivity to low-dose ACTH administration in depressed patients. Biological Psychiatry
Lazarus JH, McGregor AM, Ludgate M, Darke C, Creagh FM, Kingswood CJ (1986) Effect of
lithium carbonate therapy on thyroid immune status in manic depressive patients: a
prospective study. Journal of Affective Disorders 11:155-160.
Lesch KP, Laux G, Erb A, Pfuller H, Beckmann H (1987a) Attenuated growth hormone response
to growth hormone-releasing hormone in major depressive disorder. Biological
Psychiatry 22:1495-1499.
Lesch KP, Laux G, Pfuller H, Erb A, Beckmann H (1987b) Growth hormone (GH) response to
GH-releasing hormone in depression. Journal of Clinical Endocrinology & Metabolism
Levine J (2000) The Hypothalamus as a Major Integrating Center. In: Neuroendocrinology in
Physiology and Medicine (Conn P, Freeman, ME, ed), pp 75-95. Totowa, New Jersey:
Linkowski P, Mendlewicz J, Leclercq R, Brasseur M, Hubain P, Golstein J, Copinschi G, Van
Cauter E (1985) The 24-hour profile of adrenocorticotropin and cortisol in major
depressive illness. Journal of Clinical Endocrinology & Metabolism 61:429-438.
Loosen PT, Prange AJ, Jr. (1982) Serum thyrotropin response to thyrotropin-releasing hormone
in psychiatric patients: a review. American Journal of Psychiatry 139:405-416.
Lovenberg TW, Liaw CW, Grigoriadis DE, Clevenger W, Chalmers DT, De Souza EB,
Oltersdorf T (1995) Cloning and characterization of a functionally distinct corticotropin-
releasing factor receptor subtype from rat brain [published erratum appears in Proc Natl
Acad Sci U S A 1995 Jun 6;92(12):5759]. Proceedings of the National Academy of
Sciences of the United States of America 92:836-840.
Mann JJ, McBride PA, Malone KM, DeMeo M, Keilp J (1995) Blunted serotonergic
responsivity in depressed inpatients. Neuropsychopharmacology 13:53-64.
Marangell LB, George MS, Callahan AM, Ketter TA, Pazzaglia PJ, L'Herrou TA, Leverich GS,
Post RM (1997a) Effects of intrathecal thyrotropin-releasing hormone (protirelin) in
refractory depressed patients. Archives of General Psychiatry 54:214-222.
Marangell LB, Ketter TA, George MS, Pazzaglia PJ, Callahan AM, Parekh P, Andreason PJ,
Horwitz B, Herscovitch P, Post RM (1997b) Inverse relationship of peripheral
thyrotropin-stimulating hormone levels to brain activity in mood disorders. American
Journal of Psychiatry 154:224-230.
Mendlewicz J, Linkowski P, Kerkhofs M, Desmedt D, Golstein J, Copinschi G, Van Cauter E
(1985) Diurnal hypersecretion of growth hormone in depression. Journal of Clinical
Endocrinology & Metabolism 60:505-512.
Midgley AR, Jr., Jaffe RB (1971) Regulation of human gonadotropins. X. Episodic fluctuation of
LH during the menstrual cycle. Journal of Clinical Endocrinology & Metabolism 33:962-
Molchan SE, Hill JL, Martinez RA, Lawlor BA, Mellow AM, Rubinow DR, Bissette G,
Nemeroff CB, Sunderland T (1993) CSF somatostatin in Alzheimer's disease and major
depression: relationship to hypothalamic-pituitary-adrenal axis and clinical measures.
Psychoneuroendocrinology 18:509-519.
Muller EE, Brambilla F, Cavagnini F, Peracchi M, Panerai A (1974) Slight effect of L-
tryptophan on growth hormone release in normal human subjects. Journal of Clinical
Endocrinology & Metabolism 39:1-5.
Myers DH, Carter RA, Burns BH, Armond A, Hussain SB, Chengapa VK (1985) A prospective
study of the effects of lithium on thyroid function and on the prevalence of antithyroid
antibodies. Psychological Medicine 15:55-61.
Nemeroff CB (1988) The role of corticotropin-releasing factor in the pathogenesis of major
depression. Pharmacopsychiatry 21:76-82.
Nemeroff CB (1999) The preeminent role of early untoward experience on vulnerability to major
psychiatric disorders: the nature-nurture controversy revisited and soon to be resolved
[news; comment]. Molecular Psychiatry 4:106-108.
Nemeroff CB, Bissette G, Akil H, Fink M (1991) Neuropeptide concentrations in the
cerebrospinal fluid of depressed patients treated with electroconvulsive therapy.
Corticotrophin-releasing factor, beta-endorphin and somatostatin. British Journal of
Psychiatry 158:59-63.
Nemeroff CB, Krishnan KR, Reed D, Leder R, Beam C, Dunnick NR (1992) Adrenal gland
enlargement in major depression. A computed tomographic study [see comments].
Archives of General Psychiatry 49:384-387.
Nemeroff CB, Owens MJ, Bissette G, Andorn AC, Stanley M (1988) Reduced corticotropin
releasing factor binding sites in the frontal cortex of suicide victims. Archives of General
Psychiatry 45:577-579.
Nemeroff CB, Simon JS, Haggerty JJ, Jr., Evans DL (1985) Antithyroid antibodies in depressed
patients. American Journal of Psychiatry 142:840-843.
Nemeroff CB, Youngblood WW, Manberg PJ, Prange AJ, Jr., Kizer JS (1983) Regional brain
concentrations of neuropeptides in Huntington's chorea and schizophrenia. Science
Nestler, E., Hyman, SE., Malenka, RC (2001). Molecular Neuropharmacology: A foundation for
clinical neuroscience, McGraw Hill
: Page 280-290.
Nicholas L, Dawkins K, Golden RN (1998) Psychoneuroendocrinology of depression. Prolactin.
Psychiatric Clinics of North America 21:341-358.
O'Hara MW, Zekoski EM, Philipps LH, Wright EJ (1990) Controlled prospective study of
postpartum mood disorders: comparison of childbearing and nonchildbearing women.
Journal of Abnormal Psychology 99:3-15.
Post RM, Gold P, Rubinow DR, Ballenger JC, Bunney WE, Jr., Goodwin FK (1982) Peptides in
the cerebrospinal fluid of neuropsychiatric patients: an approach to central nervous
system peptide function. Life Sciences 31:1-15.
Prange AJ (1996) Novel uses of thyroid hormones in patients with affective disorders. Thyroid
Prange AJ, Jr., Lara PP, Wilson IC, Alltop LB, Breese GR (1972) Effects of thyrotropin-
releasing hormone in depression. Lancet 2:999-1002.
Primus RJ, Yevich E, Baltazar C, Gallager DW (1997) Autoradiographic localization of CRF1
and CRF2 binding sites in adult rat brain. Neuropsychopharmacology 17:308-316.
Richardson UI, Schonbrunn A (1981) Inhibition of adrenocorticotropin secretion by somatostatin
in pituitary cells in culture. Endocrinology 108:281-290.
Risch S (1991) Growth hormone-releasing factor and growth hormone. In: Neuropeptides and
Psychiatric Disorders (Nemeroff C, ed), pp 93-108. Washington DC: American
Psychiatric Press.
Risch SC, Lewine RJ, Kalin NH, Jewart RD, Risby ED, Caudle JM, Stipetic M, Turner J, Eccard
MB, Pollard WE (1992) Limbic-hypothalamic-pituitary-adrenal axis activity and
ventricular-to-brain ratio studies in affective illness and schizophrenia.
Neuropsychopharmacology 6:95-100.
Roy A, Pickar D, Paul S, Doran A, Chrousos GP, Gold PW (1987) CSF corticotropin-releasing
hormone in depressed patients and normal control subjects. American Journal of
Psychiatry 144:641-645.
Ryan ND, Dahl RE, Birmaher B, Williamson DE, Iyengar S, Nelson B, Puig-Antich J, Perel JM
(1994) Stimulatory tests of growth hormone secretion in prepubertal major depression:
depressed versus normal children. Journal of the American Academy of Child &
Adolescent Psychiatry 33:824-833.
Sachar EJ, Hellman L, Fukushima DK, Gallagher TF (1970) Cortisol production in depressive
illness. A clinical and biochemical clarification. Archives of General Psychiatry 23:289-
Sack DA, James SP, Rosenthal NE, Wehr TA (1988) Deficient nocturnal surge of TSH secretion
during sleep and sleep deprivation in rapid-cycling bipolar illness. Psychiatry Research
Sadock B, Sadock, V. (2000) Kaplan & Sadock's comprehensive textbook of psychiatry, VII
Edition. Philadelphia: Lippincott Williams & Williams.
Saffran M, Schally, AV., Benfey, BG (1955) Stimulation of the release of corticotropin from the
adenohypophysis by a neruhypophysial factor. Endocrinology 57:439-444.
Sanchez MM, Young LJ, Plotsky PM, Insel TR (1999) Autoradiographic and in situ
hybridization localization of corticotropin-releasing factor 1 and 2 receptors in nonhuman
primate brain. Journal of Comparative Neurology 408:365-377.
Schilkrut R, Chandra O, Osswald M, Ruther E, Baafusser B, Matussek, N. (1975) Growth
hormone release during sleep and with thermal stimulation in depressed patients.
Neuropsychobiology 1:70-79.
Shaw C (1892) Case of myxedema with restless melancholia treated by injections of thyroid
juice:recovery. British Journal of Medicine 451.
Siever LJ, Uhde TW, Silberman EK, Jimerson DC, Aloi JA, Post RM, Murphy DL (1982a)
Growth hormone response to clonidine as a probe of noradrenergic receptor
responsiveness in affective disorder patients and controls. Psychiatry Research 6:171-
Siever LJ, Uhde TW, Silberman EK, Lake CR, Jimerson DC, Risch SC, Kalin NH, Murphy DL
(1982b) Evaluation of alpha-adrenergic responsiveness to clonidine challenge and
noradrenergic metabolism in the affective disorders and their treatment.
Psychopharmacology Bulletin 18:118-119.
Skare SS, Dysken MW, Billington CJ (1994) A review of GHRH stimulation test in psychiatry.
Biological Psychiatry 36:249-265.
Souetre E, Salvati E, Wehr TA, Sack DA, Krebs B, Darcourt G (1988) Twenty-four-hour
profiles of body temperature and plasma TSH in bipolar patients during depression and
during remission and in normal control subjects. American Journal of Psychiatry
Spillane J (1951) Nervous and mental disorders in Cushing's syndrome. Brain 74:72-94.
Suda T, Tomori N, Tozawa F, Mouri T, Demura H, Shizume K (1984) Distribution and
characterization of immunoreactive corticotropin-releasing factor in human tissues.
Journal of Clinical Endocrinology & Metabolism 59:861-866.
Swanson LW, Sawchenko PE, Rivier J, Vale WW (1983) Organization of ovine corticotropin-
releasing factor immunoreactive cells and fibers in the rat brain: an immunohistochemical
study. Neuroendocrinology 36:165-186.
Unden F, Ljunggren JG, Beck-Friis J, Kjellman BF, Wetterberg L (1988) Hypothalamic-
pituitary-gonadal axis in major depressive disorders. Acta Psychiatrica Scandinavica
Vale W, Spiess J, Rivier C, Rivier J (1981) Characterization of a 41-residue ovine hypothalamic
peptide that stimulates secretion of corticotropin and beta-endorphin. Science 213:1394-
Van de Kar LD (1989) Neuroendocrine aspects of the serotonergic hypothesis of depression.
Neuroscience & Biobehavioral Reviews 13:237-246.
Van Pett K, Viau, V., Bittencourt, JC., Chan RK., Li, HY.,Arias, C., Prins GS., Perrin M., Vale
W., Sawchenko PE. (2000) Distribution of mRNAs encoding CRF receptors in brain and
pituitary of rat and mouse. Journal of Comparative Neurology 428:191-212.
Vaughan J, Donaldson C, Bittencourt J, Perrin MH, Lewis K, Sutton S, Chan R, Turnbull AV,
Lovejoy D, Rivier C (1995) Urocortin, a mammalian neuropeptide related to fish
urotensin I and to corticotropin-releasing factor [see comments]. Nature 378:287-292.
Veith RC, Lewis N, Langohr JI, Murburg MM, Ashleigh EA, Castillo S, Peskind ER, Pascualy
M, Bissette G, Nemeroff CB (1993) Effect of desipramine on cerebrospinal fluid
concentrations of corticotropin-releasing factor in human subjects. Psychiatry Research
Veldhuis D (2000) The Neuroendocrine Control of Ultradian Rhythms. In: Neuroendocrinology
in Physiology and Medicine (Conn P, Freeman, ME, ed), pp 453-475. Totowa, New
Jersey: Humana Press.
Whybrow PC, Prange AJ, Jr., Treadway CR (1969) Mental changes accompanying thyroid gland
dysfunction. A reappraisal using objective psychological measurement. Archives of
General Psychiatry 20:48-63.
Winokur A, Amsterdam J, Caroff S, Snyder PJ, Brunswick D (1982) Variability of hormonal
responses to a series of neuroendocrine challenges in depressed patients. American
Journal of Psychiatry 139:39-44.
Wisner KL, Stowe ZN (1997) Psychobiology of postpartum mood disorders. Seminars In
Reproductive Endocrinology 15:77-89.
Wynn PC, Aguilera G, Morell J, Catt KJ (1983) Properties and regulation of high-affinity
pituitary receptors for corticotropin-releasing factor. Biochemical & Biophysical
Research Communications 110:602-608.
Wynn PC, Harwood JP, Catt KJ, Aguilera G (1988) Corticotropin-releasing factor (CRF)
induces desensitization of the rat pituitary CRF receptor-adenylate cyclase complex.
Endocrinology 122:351-358.
Wynn PC, Hauger RL, Holmes MC, Millan MA, Catt KJ, Aguilera G (1984) Brain and pituitary
receptors for corticotropin releasing factor: localization and differential regulation after
adrenalectomy. Peptides 5:1077-1084.
Yehuda R, Teicher MH, Trestman RL, Levengood RA, Siever LJ (1996) Cortisol regulation in
posttraumatic stress disorder and major depression: a chronobiological analysis.
Biological Psychiatry 40:79-88.
Young EA, Midgley AR, Carlson NE, Brown MB (2000) Alteration in the hypothalamic-
pituitary-ovarian axis in depressed women. [see comments]. Archives of General
Psychiatry 57:1157-1162.
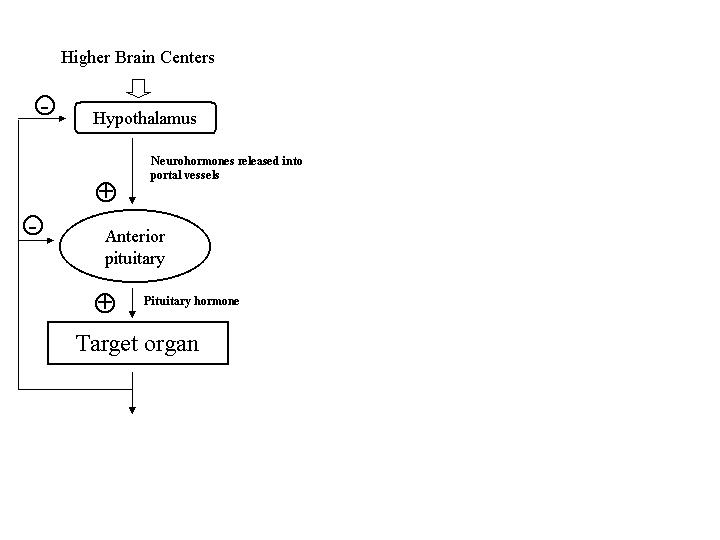
Young EA, Watson SJ, Kotun J, Haskett RF, Grunhaus L, Murphy-Weinberg V, Vale W, Rivier
J, Akil H (1990) Beta-lipotropin-beta-endorphin response to low-dose ovine corticotropin
releasing factor in endogenous depression. Preliminary studies. Archives of General
Psychiatry 47:449-457.
Zeiger MA, Fraker DL, Pass HI, Nieman LK, Cutler GB, Jr., Chrousos GP, Norton JA (1993)
Effective reversibility of the signs and symptoms of hypercortisolism by bilateral
adrenalectomy. Surgery 114:1138-1143.
Zobel AW, Nickel T, Kunzel HE, Ackl N, Sonntag A, Ising M, Holsboer F (2000) Effects of the
high-affinity corticotropin-releasing hormone receptor 1 antagonist R121919 in major
depression: the first 20 patients treated. Journal of Psychiatric Research 34:171-181.
Figure 1. Overview of the common organizational motif of the neuroendocrine axis. The neurosecretion of hypothalamic factors into hypophyseal portal vessels is regulated by a set point of activity from higher brain centers. Neurohormones released from the hypothalamus into hypophyseal portal vessels in turn stimulate cells in the pituitary. These adenohypophyseal hormones then regulate the hormone output from the end-organ. The end organ then exerts negative feedback effects at the pituitary and hypothalamus to prevent further neurohormone and pituitary hormone release via "long-loop" negative feedback. Short-loop negative feedback may also occur where pituitary hormones feed back directly on hypothalamic neurons to prevent further neurohormone release.
Figure 2. Diagram of the neurovascular anatomy of the hypothalamic-pituitary axis. PVN, paraventricular nucleus. SON, supraoptic nucleus. POA, preoptic area. ARC, arcuate nucleus. PT, pars tuberalis. PI, pars intermedia. PD, pars distalis. PN, pars nervosa, MB, Mamillary Body, OC, Optic chiasm.
Figure 3- Overview of the feedback system of the Hypothalamic-Pituitary-Thyroid (HPT) axis. Thyroid-releasing hormone (TRH) from the hypothalamus stimulates TSH from the pituitary, which stimulates thyroid hormone release. As circulating thyroid hormone levels increase, they inhibit further release of TSH and TRH. Other hypothalamic-pituitary-end organ axes exhibit similar feedback control mechanisms.
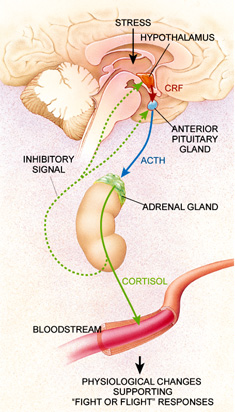
Figure 4- Overview of the feedback mechanisms of the hypothalamic-pituitary-adrenal axis. Following relevant stimuli,
including stress, corticotropin-releasing factor (CRF) is released from the hypothalamus into hypophyseal portal vessels where it
is transported in high concentrations to the pituitary gland. CRF then promotes the release of adrenocorticotropin (ACTH),
which in turn promotes the release of cortisol from the adrenal glands. Cortisol acts as an inhibitory signal at both the
hypothalamus and pituitary preventing further CRF and ACTH release, respectively. Mounting evidence suggests that chronic
overactivity of the axis, and particularly overproduction of corticotropin-releasing factor (CRF), may contribute to the
pathophysiology of depression. Reproduced from (Nemeroff, CB 1998) with permission.
Table 1: List of major releasing factors and their hormonal target Neurohormone/Releasing Factor
Hormone Stimulated
Corticotropin-releasing factor (CRF)
Adrenocorticotropic hormone (ACTH)
Thyrotropin-releasing hormone (TRH) Thyroid-stimulating
Gonadotropin-releasing hormone (GnRH)
Follicle-stimulating hormone (FSH)
Somatostatin (SRIF)
Growth Hormone (GH)
Growth-hormone-releasing hormone (GHRH)
Arginine vasopressin (AVP)
Oxytocin Prolactin
Table 2: HPA Axis changes demonstrated in depression ↑ Corticotropin-releasing factor (CRF) in cerebrospinal fluida
↓ Adrenocorticotropic hormone response (ACTH) to CRF stimulation ↓ Density of CRF receptors in frontal cortex of suicide victims Enlarged pituitary gland in depressed patients a Adrenal gland enlargement in suicide victims and depressed patients ↑ Plasma cortisol during depression a ↑ Urinary free cortisol concentrations a Nonsuppression of plasma cortisol and ACTH after dexamethasone administration a a State-dependent
Table 3: Hypothalamic-pituitary-thyroid (HPT) axis alterations in depression ↑ CSF TRH in depressed patients
↓ Nocturnal plasma TSH Blunted TSH in response to TRH stimulation (state dependent) Exaggerated TSH response to TRH stimulation ↓ ΔΔTSH (difference between 11 P.M. ΔTSH and 8 A.M. ΔTSH after TRH administration) Presence of antithyroglobulin and/or antimicrosomal thyroid antibodies
Table 4: Growth hormone (GH) axis changes in depression ↑ Circulating daily GH levels (uni- and bipolar depression) ↓ Noctural GH in depression
↓ Response of GH to clonidine
INDEX WORDS: Hypothalamic-Pituitary-Axis HPA Axis CRF Stimulation Test Corticotropin-Releasing Factor Corticotropin-Releasing Hormone Cortisol Hypercortisolemia Adrenocorticotropin Hormine (ACTH) Glucocorticoids Cortisol and depression CRF receptors Cushing's Syndrome Dexamethasone Suppression Test (DEX Test) DEX/CRF Test Trier Social Stress Test (TSST) HPT Axis (Hypothalamic-Pituitary-Thyroid Axis) Thyroid T3 and T4
TSH (Thyroid Stimulating Hormone) TRH (Thyrotropin Releasing Hormone) Hypothyroidism Hyperthyroidism Hypothyroidism and Depression HPG Axis (Hypothalamic-Pituitary-Gonadal Axis) Follicle Stimulating Hormone (FSH) Luteinizing Hormone (LH) Postpartum Depression Hypothalamic-Prolactin Axis Prolactin Hyperprolactinemia Oxytocin Vasopressin Growth Hormone (GH) Growth-Hormone-Releasing Factor (GHRF) Growth-Hormone-Inhibiting Hormone (GHIH) GH response to clonidine GHRH Stimulation Test
Source: http://www.drgutman.org/CV/ORIGINAL_PAPERS/neuroendocrinology_textbook_biological_psych.pdf
Scientific Committee Teresa Giraldez (Hospital Universitario Ntra. Sra. de Candelaria, Tenerife, Spain) Luis A. Pardo (Max Planck Institute for Experimental Medicine, Göttingen, Germany)
MINISTÈRE DES AFFAIRES ÉTRANGÈRES & LE COMITÉ D'INFORMATION MÉDICALE (Ministère des Affaires Étrangères – Maison des Français à l'Étranger) Mémento à l'usage des professionnels de santé La Société de Médecine des VoyagesetLe Comité d'Information MédicaleMinistère des Affaires Étrangères –Maison des Français de l'Étranger EXPATRIATION ET SANTÉ,Mémento à l'usage des professionnels de santé