Print
Clin Genet 2010: 78: 471–477
2010 John Wiley & Sons A/S
Printed in Singapore. All rights reserved
Short Report
Refining the phenotype associated with
MEF2C haploinsufficiency
Novara F, Beri S, Giorda R, Ortibus E, Nageshappa S, Darra F,
F Novaraa, S Berib, R Giordab,
dalla Bernardina B, Zuffardi O, Van Esch H. Refining the phenotype
E Ortibusc, S Nageshappad,
associated with
MEF2C haploinsufficiency.
F Darrae, B dalla Bernardinae,
Clin Genet 2010: 78: 471–477. John Wiley & Sons A/S, 2010
O Zuffardia,f and H Van Eschd
Recently, submicroscopic deletions of the 5q14.3 region have been
aGenetica Medica, Universit a di Pavia,
described in patients with severe mental retardation (MR), stereotypic
Pavia, 27100 PV, Italy, bBiologiaMolecolare, IRCCS ‘‘E. Medea'', Bosisio
movements, epilepsy and cerebral malformations. Further delineation of a
Parini, Lecco, Italy, cDepartment of
critical region of overlap in these patients pointed to
MEF2C as the
Pediatrics and dCenter for Human
responsible gene. This finding was further reinforced by the identification
Genetics, University Hospitals Leuven,
of a nonsense mutation in a patient with a similar phenotype. In brain,
Leuven, Belgium, eNeuropsichiatria
MEF2C is essential for early neurogenesis, neuronal migration and
Infantile, Policlinico GB Rossi, Verona,
differentiation. Here we present two additional patients with severe MR,
Italy, and fIRCCS Fondazione C.
autism spectrum disorder and epilepsy, carrying a very small deletion
Mondino, Pavia, Italy
encompassing the
MEF2C gene. This finding strengthens the role of this
Key words: aCGH – chromosome
gene in severe MR, and enables further delineation of the clinical
5q14.3 – epilepsy – haploinsufficiency –MEF2C – microdeletion – severe mental
Corresponding author: Hilde Van Esch,MD, PhD, Centre for Human Genetics,University Hospitals Leuven, Herestraat49, 3000 Leuven, Belgium.
Tel.: +32 16 345903;fax: +32 16 346051;e-mail: Hilde.Vanesch@med.
kuleuven.be
Received 6 November 2009, revisedand accepted for publication 22February 2010
Recently, submicroscopic deletions of the 5q14.3
factors are expressed in overlapping but distinct
region have been described in patients featuring
regions of the central nervous system (CNS) that
severe mental retardation (MR), stereotypic move-
correlate with the withdrawal of neurons from
ments, epilepsy and cerebral malformations (1–3).
the cell cycle and acquisition of a differentiated
Further delineation of a critical region of overlap
phenotype (4). In mouse,
Mef2c is the first of
in these patients pointed to the
MEF2C gene as
four
Mef2 genes to be expressed in the CNS. In
the responsible gene. This finding was further rein-
the adult brain,
Mef2c is highly expressed in the
forced by the identification of a nonsense mutation
frontal cortex, entorhinal cortex, dentate gyrus, and
in a patient with a similar phenotype.
MEF2C,
amygdala. Recently it was shown that the dele-
encoding transcription factor myocyte enhancer
tion of
Mef2c transcription factor in the CNS of
factor 2C, plays a crucial role during several
mice impairs hippocampal-dependent learning and
embryological processes, including hematopoiesis,
memory by negative regulation of synapse num-
cardiogenesis and neurogenesis. In brain, members
bers and function (5, 6).
of the MEF2 family of MADS (MCM1, agamous,
Here we present two additional patients with
deficiens, serum response factor) box transcription
severe MR, autism spectrum disorder and epilepsy,
Novara et al.
carrying a very small deletion encompassing the
Milan, Italy) with the following protocol: 30 s at
MEF2C gene. This finding strengthens the role
96◦C, 35 cycles of 15 s at 94◦C/20 s at 58◦C/10
of this gene in severe MR, and enables further
min at 68◦C, 10 min final elongation time. Primers
delineation of the clinical phenotype.
were: for Patient 1, Del5-11F (5-CATCATTGCCCCACATCACA-3) and Del5-13R (5-TGAAGGAGAGCTGGCTGTGA-3); for Patient 2, Del5-10F
(5-TGTGGCTGAGCTGCTTCTAACA-3) and Del
The protocol was approved by the appropri-
5-11R (5-TTCCTGCCCTACCTTCATGTG-3). All
ate Institutional Review Boards involved in the
sequencing reactions were performed with a Big
research (Universities of Leuven, Belgium and
Dye Terminator Cycle Sequencing kit 3.1 (Applied
Pavia, Italy). Informed consent was obtained from
Biosystems) and run on an ABI Prism 3130xl
the parents of the affected patients.
Genetic Analyzer.
The UCSC Genome Browser (May 2004 assem-
bly; http://genome.ucsc.edu/) maps and sequence
Cytogenetic analysis
were used as references. The primer sequences are
available on request.
according to routine protocol. Arrays were per-formed using the Agilent array 105 K according
to the manufacturer's protocol.
Patient 1 is the first child of healthy non-relatedparents, born at term with normal birth parame-
ters after an uneventful pregnancy. Two younger
Genotyping of polymorphic loci in patient 2 and
siblings are normal. A brother and a cousin of
his parents was performed by amplification with
the mother have a benign form of epilepsy, well
primers labeled with fluorescent probes (ABI
responding to therapy and not interfering with
6-Fam and 8-Hex), followed by analysis on ABI
daily functioning or cognition. The boy came to
3100 Genetic Analyzer (Applied Biosystems, Fos-
medical attention at the age of 3 months because
ter City, CA). Primers were designed using the
of absent eye contact and social smile, hypoto-
database tool Tandem Repeats Finder (http://
nia and irritable behavior. MRI scan at that age
showed a cystic lesion and leucoencephalopathyin the left frontal region, probably due to a peri-natal hemorrhage. Metabolic investigations includ-
Real-time quantitative PCR
ing coagulation were all normal. Subsequently, his
Specific target sequences were selected for Real-
psychomotor development was severely delayed
time quantitative PCR (qPCR) using Primer
with sitting with little support at the age of 2 years.
Express software (Applied Biosystems). A control
An MRI scan of the brain at that age showed signs
amplicon was selected with the same parameters
of periventricular leucomalacia and atrophy of the
in the
MAPK1 gene on 22q11.2; size (approxi-
frontal cortex at the left side. He never reached
mately 60 bp) and Tm (59◦C) were the same for all
independent ambulation because of persistent
amplicons. Amplification and detection were per-
severe axial hypotonia. Epilepsy started in the first
formed on an ABI PRISM 7700 Sequence Detec-
year of life, initially as isolated myoclonic jerks,
tion System (Applied Biosystems) using SYBR
later evolving toward infantile spasms with con-
Green PCR Master Mix (Applied Biosystems);
tinuous epileptic activity bi-posterior with no basic
thermal cycling conditions were 50◦C for 2 min
rhythm on EEG. Because of further deterioration
and 95◦C for 10 min, followed by 40 cycles of
of the drug-resistant epilepsy, he was admitted at
95◦C for 15 s and 60◦C for 1 min; all samples
a special epilepsy unit for intensive care at the age
were amplified in duplicate. Relative quantifica-
of 2 years. After this period there was a regres-
tion of the amount of DNA was obtained using
sion of his motor functions, necessitating a wheel
the comparative CT method (described in Applied
chair and orthopedic ortheses. Speech was absent.
Biosystems User Bulletin #2, December 11, 1997:
Now at the age of 14 years, his growth parameters
ABI PRISM 7700 Sequence Detection System).
are within the normal range (OFC at 50th centile).
He has a cerebral palsy with severe axial hypoto-nia and compensatory peripheral hypertonia. His
eye contact is very poor with external strabismus
Long-range PCRs were performed with Jump-
of the right eye. He does not exhibit stereotypic
Start Red ACCUTaq LA DNA polymerase (Sigma,
movements. He has only mild dysmorphisms with
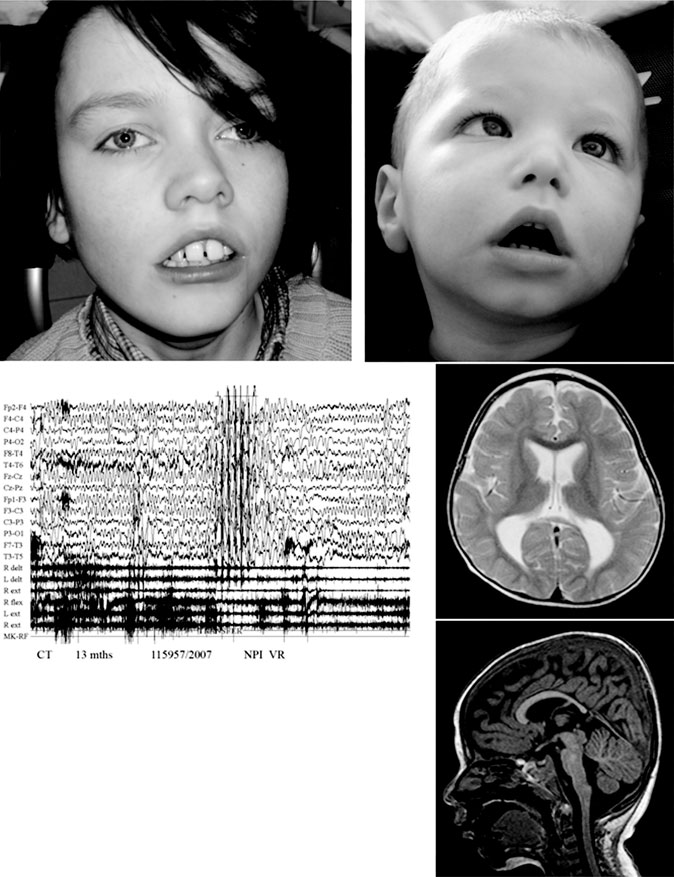
Deletion of MEF2C causes severe MR
Fig. 1. Clinical picture of patient 1 at the age of 14 years (a) and patient 2 at the age of 3 years (b). Note the facial hypotonia,
strabismus, prominent philtrum with cupid's bow in both boys. Patient 1 has in addition macrodontia. (c) EEG record of patient 2 at
the age of 13 months showing slow background activity with theta waves degraded over the central regions of the two hemispheres
and degraded diffuse discharges, together with rhythmic sharp wave activity; EEG velocity: 15 mm/s; amplitude: 50 μV. (d,e) MRI
images of patient 2: axial section showing dilatation of the lateral ventricles (d) and sagittal section showing hypoplasia of the
distal part of the corpus callosum (e).
prominent ear lobes, short prominent philtrum with
His mother suffered from hypothyroidism and was
a cupid's bow and macrodontia (Fig. 1a).
treated with levothyroxin during pregnancy. At
Patient 2 was born at term with birth weight of
4 months of age, lack of reactivity was observed
3600 g and length of 51 cm (both 75th centile).
in addition to severe hypotonia, dystonic motor
Novara et al.
activity, absent head control and poor visual track-
(patient 2) mechanisms. (Table S1, Supporting
ing. At the age of 5 months, psychomotor delay
information). Parental origin was not investigated
and myoclonus were observed. EEG showed slow
in patient 1. In patient 2, the chromosomal imbal-
background activity with theta waves degraded
ance originated at the paternal meiosis. Three out
over the central regions of the two hemispheres
of four microsatellites analyzed in the deleted
and degraded diffuse discharges, sometimes with
region showed the presence of the maternal allele
episodes of rhythmic sharp wave activity, asso-
only (data not shown).
ciated with revulsion of eyes and myoclonus ofthe limbs (Fig. 1c). Clinical evaluation showed
occipital plagiocephaly, hypertelorism, flattenednasal bridge, small and hooked nose, ogival
Recently, MEF2C haploinsufficiency has been
palate, and low-set and dysmorphic ears (Fig. 1b).
described in five patients presenting a severe and
Marked myopia with alternating esotropia was also
syndromic form of MR and carrying a submicro-
observed. Several MRI scans of the brain were
scopic deletion on chromosome 5q14.3 of variable
performed at 5, 8 and 19 months of age, respec-
size ranging from 216 kb to 8.8 Mb (1). Moreover,
tively; they showed moderate dilatation of lateral
the same authors detected a nonsense mutation
ventricles and hypoplasia of the corpus callosum
in MEF2C in an additional patient exhibiting a
with abnormal aspect of the splenius (Fig. 1d,e).
similar phenotype and thus underscoring the clin-
The development quotient was calculated using the
ical relevance of this new autosomal dominant
Griffith mental development scales at 13 and 19
MR gene. MEF2C encodes myocyte enhancer
months of age (35 and 30, respectively). At the
factor 2 that functions as a transcription factor,
last evaluation at 3 years and 10 months of age,
with MEF2C as the predominant isoform in the
he presented a severe cognitive deficit with numer-
brain. The role of MEF2C during brain develop-
ous behavioral stereotypes. Head circumference
ment and functioning, including neurogenesis, neu-
was at the 50th centile. Neurological examination
ronal migration and synaptic plasticity, has been
showed severe axial hypotonia with partial control
well established in murine models and in vitro
of the head. In the lower limbs an increased mus-
functional studies (4–7). Its role in learning and
cular tonus was noticed with dystonic–dyskinetic
memory as well as maintaining the critical bal-
movements. He was able to fix and follow objects
ance between inhibitory and excitatory synapses
and wore glasses for myopia. Language was
is consistent with the human haploinsufficiency
absent. He still suffered from myoclonic and
phenotype we and others have observed. Both
myoclonic–atonic seizures with falling of the
patients we describe here carry a very small dele-
head, not responding to any anti-epileptic ther-
tion, involving only the MEF2C in patient 1. The
apy (Valproic Acid, Nitrazepam, Levetiracetam,
deletion in patient 2 also affects the TMEM161B
Hydrocortison, Clobazam, and Ethosuximide).
gene, a gene of unknown function that is predictedto encode a transmembrane protein. We can notexclude an involvement of haploinsufficiency of
this gene in the clinical phenotype of patient 2.
In the absence of an etiological diagnosis in both
When we compare the clinical phenotype in
patients, array CGH analysis using the 105 K
our patients with the reported patients, the pheno-
array (Agilent) was performed. In both patients,
type remains very consistent (Table 1). All patients
a small deletion in 5q14.3 was detected (Fig. 2).
present with severe primary developmental delay
All breakpoint locations were refined by qPCR,
reflected by early hypotonia, delayed motor devel-
and then both junctions were amplified by long-
opment and no speech development. Thus far, our
range PCR and sequenced. Patient 1 has a deletion
patients and previously reported patients who are
of 318357 bp (87,978,527–88,296,884) harboring
hemizygous for the gene did not reach indepen-
only MEF2C, while patient 2 has a slightly larger
dent walking and remain very hypotonic, while
deletion of 1140131 bp (87,234,127–88,374,258).
the patient with a reported point mutation started
Besides MEF2C, the latter deletion also includes
to walk at 3 years of age without hypotonia (1)
the TMEM161B gene. The mother of patient 1
(Table 1). This might point toward a more severe
does not carry the deletion, while his father could
effect of the deletion compared to the intragenic
not be tested. In patient 2 the deletion occurred
mutation, but more cases are needed to make
de novo. Breakpoint junctions are outside low
any genotype–phenotype correlation. Stereotypic
copy repeats and have no particular identity.
movements and poor eye contact are present in
They are consistent with microhomology-mediated
many patients, suggesting the diagnosis of autism
(patient 1) or non-homologous end-joining (NHEJ)
spectrum disorder (ASD). Interestingly, a role for

Deletion of MEF2C causes severe MR
Fig. 2. Detailed view of the chromosome 5q14.3 aCGH profile showing the deletion in patient 1 (right) and patient 2 (left).
MEF2C in ASD was already shown in classical
is very distinct in severity, we believe that another
and conditional knock-out mouse models (4, 8).
genetic trait is reponsible for their epilepsia.
Moreover, Morrow et al. identified many MEF2
In most patients brain imaging is reported to
target genes in their screen for autism genes by
be abnormal, including anomalies of the cor-
means of homozygosity mapping in pedigrees with
pus callosum, enlarged ventricles, periventricular
shared ancestry (9). The recent findings in humans
white matter hyperintensities and cortical atrophy
further reinforce the role of MEF2C during neu-
(Table 1). None of these anomalies seems to be
rogenesis and synaptogenesis. Epilepsy is another
specific and some might be secondary to the severe
frequent feature of MEF2C haploinsufficiency,
epileptic activity. Interestingly, Cardoso et al.
although the type (myoclonic, tonic-clonic, infan-
reported periventricular heterotopia in a patient
tile spasms and febrile seizures) and age at onset
with a larger deletion including MEF2C (2). For
may vary considerably. In both patients the severe
the moment it is unclear whether MEF2C haploin-
drug-resistant infantile spasms even necessitated
sufficiency is responsible for this variable spectrum
admission at an epilepsy care center. Patient 1 has
of features or whether other genes within larger
two male relatives with a benign form of epilepsy,
deletions exert an additional effect. The same holds
not affecting cognition or daily functioning. As the
true for the facial dysmorphisms that seem to
deletion is de novo, and the phenotype in patient 1
be more pronounced in the patients with larger
Novara et al.
Deletion of MEF2C causes severe MR
deletions (1). We did not observe any effect of
MEF2C hemizygosity on growth and head circum-
1. Le Meur N, Holder-Espinasse M, Jaillard S et al. MEF2C hap-
ference (Table 1).
loinsufficiency caused either by microdeletion of the 5q14.3
In summary, we present two new patients with
region or mutation is responsible for severe mental retardation
severe MR, epilepsy and ASD associated with
with stereotypic movements, epilepsy and/or cerebral malfor-
deletion of MEF2C.
mations. J Med Genet 2009: 47: 22 – 29.
2. Cardoso C, Boys A, Parrini E et al. Periventricular heterotopia,
mental retardation, and epilepsy associated with 5q14.3 – q15deletion. Neurology 2009: 72: 784 – 792.
3. Engels H, Wohlleber E, Zink A et al. A novel microdeletion
The following Supporting information is available for this article:
syndrome involving 5q14.3 – q15: clinical and molecular cyto-
Table S1. Cloning of the deletion breakpoints in patients 1 and 2.
genetic characterization of three patients. Eur J Human Genetics2009: 17: 1592 – 1599.
Additional Supporting information may be found in the online
4. Li H, Radford JC, Ragusa MJ et al. Transcription factor
version of this article.
MEF2C influences neural stem/progenitor cell differentiation
Please note: Wiley-Blackwell Publishing is not responsible for the
and maturation in vivo. Proc Natl Acad Sci U S A 2008: 105:
content or functionality of any supplementary materials supplied
9397 – 9402.
by the authors. Any queries (other than missing material) should
5. Barbosa AC, Kim MS, Ertunc M et al. MEF2C, a transcription
be directed to the corresponding author for the article.
factor that facilitates learning and memory by negative regu-lation of synapse numbers and function. Proc Natl Acad SciU S A 2008: 105: 9391 – 9396.
6. Flavell SW, Cowan CW, Kim TK et al. Activity-dependent
regulation of MEF2 transcription factors suppresses excitatory
We thank the families for their cooperation. H.V.E. is funded by
synapse number. Science 2006: 311: 1008 – 1012.
the F.W.O. Vlaanderen. O.Z. is funded by Cassa di Risparmio
7. Shalizi A, Gaudilliere B, Yuan Z et al. A calcium-regulated
delle Provincie Lombarde (CARIPLO: 2007.5197, bando 2007) and
MEF2 sumoylation switch controls postsynaptic differentiation.
by Ministry of Health Grant (RF-AOM-2007-636538. ‘Genomic
Science 2006: 311: 1012 – 1017.
structural variation studies in mentally retarded and normal
8. Lipton SA, Li H, Zaremba JD et al. Autistic phenotype from
individuals in Italy').
MEF2C knockout cells. Science 2009: 323: 208.
9. Morrow EM, Yoo SY, Flavell SW et al. Identifying autism loci
and genes by tracing recent shared ancestry. Science 2008: 321:
Conflict of interest
218 – 223.
The authors do not have any affiliation with anygroup with a direct financial interest in the subjectmatter or materials discussed in the manuscript.
Copyright of Clinical Genetics is the property of Wiley-Blackwell and its content may not be copied or emailed
to multiple sites or posted to a listserv without the copyright holder's express written permission. However,
users may print, download, or email articles for individual use.
Source: http://pfbc-construct.be/MEF2C/files/Case%20Publication%202010%20Novara.pdf
Fireweed Toxicity Facts and Steven M Colegate BSc(Hons), PhD Photos: Forest&Kim Starr Fireweed Toxicity Facts and Perspectives Executive Summary • HPLC-MS analysis of fireweed collected in the Bega Valley (NSW) in the spring of 2006 and 2008 showed the presence of dehydropyrrolizidine alkaloid esters at levels up to about 220 milligrams per kilogram of plant.
A High Throughput Approach for Metabolite Note: 369 Profiling and Characterization Using theLXQ Linear Ion Trap Mass SpectrometerMin He, Alicia Du, Gargi Choudhary, Karen Salomon and Diane Cho; Thermo Electron Corporation, San Jose, CA, USA Key Words Within the drug discovery environment, high sample • LXQ™ throughput that provides comprehensive drug metabolite