Crystal structure of equine serum albumin in complex with cetirizine reveals a novel drug binding site



Contents lists available at
Molecular Immunology
Crystal structure of equine serum albumin in complex with cetirizine
reveals a novel drug binding site
Katarzyna B. Handing , Ivan G. Shabalin , Karol Szlachta , Karolina A. Majorek ,
a Department of Molecular Physiology and Biological Physics, University of Virginia, Charlottesville, VA 22908-0736, USA
b New York Structural Genomics Research Consortium (NYSGRC), USA
c Faculty of Physics, Warsaw University of Technology, 00-662 Warszawa, Poland
Serum albumin (SA) is the main transporter of drugs in mammalian blood plasma. Here, we report the
Received 14 December 2015
first crystal structure of equine serum albumin (ESA) in complex with antihistamine drug cetirizine at a
Received in revised form 2 February 2016
resolution of 2.1 Å. Cetirizine is bound in two sites—a novel drug binding site (CBS1) and the fatty acid
Accepted 8 February 2016
binding site 6 (CBS2). Both sites differ from those that have been proposed in multiple reports based
on equilibrium dialysis and fluorescence studies for mammalian albumins as cetirizine binding sites.
We show that the residues forming the binding pockets in ESA are highly conserved in human serum
albumin (HSA), and suggest that binding of cetirizine to HSA will be similar. In support of that hypothesis,
we show that the dissociation constants for cetirizine binding to CBS2 in ESA and HSA are identical using
tryptophan fluorescence quenching. Presence of lysine and arginine residues that have been previously
reported to undergo nonenzymatic glycosylation in CBS1 and CBS2 suggests that cetirizine transport in
Crystal structure
patients with diabetes could be altered. A review of all available SA structures from the PDB shows that
in addition to the novel drug binding site we present here (CBS1), there are two pockets on SA capable of
binding drugs that do not overlap with fatty acid binding sites and have not been discussed in published
2016 Elsevier Ltd. All rights reserved.
ever, drugs with high affinity for SA may require higher doses to
achieve the desired effect
Serum albumin (SA) is the most abundant protein in mam-
Previous studies of SA revealed several distinct drug binding
malian blood plasma. Due to its high concentration and presence
sites (Two canonical drug binding sites were
of multiple binding pockets, SA is a major transporter of endoge-
proposed by Sudlow et al. before a crystal structure of SA was avail-
nous compounds, including fatty acids, hormones, and metal ions
able (As was later confirmed by multiple
Many commonly used drugs such as warfarin,
crystallographic studies, drug site 1 (Sudlow site I) is located in
diazepam, and ibuprofen also bind to SA, which can be beneficial
subdomain IIA, and drug site 2 (Sudlow site II) in subdomain IIIA.
for drug delivery. Binding to SA can increase solubility, decrease the
Recently, that several oncological drugs
formation of aggregates, and increase the half-life of a drug. How-
bind to a site in subdomain IB, and this site was proposed to be
the third drug binding site. In addition to the main drug binding
sites, some drugs were found to bind in fatty acid binding sites
have revealed nine fatty acid sites on SA: seven that bind long-
Abbreviations: CBS, cetirizine binding site; ESA, equine serum albumin; HEPES,
chain fatty acids (FA1-7) and two for short chain fatty acids (FA8
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid; HSA, human serum albumin;
and FA9) (All three main
IFE, internal filter effect; LLDF, local ligand density fit; NEG, nonenzymatic glycosy-
lation; PBS, phosphate-buffered saline; PDB, Protein Data Bank; PEG, polyethylene
drug binding sites overlap with fatty acid binding sites: drug site 1
glycol; pIFE, primary internal filter effect; RMSD, root-mean-square deviation; RSR,
overlaps with FA7, drug site 2 with FA3/FA4, and drug site 3 with
real space R-factor; SA, serum albumin; TFQ, tryptophan fluorescence quenching;
FA1. Therefore, all FA binding sites are considered as potential drug
TLS, translation/libration/screw; sIFE, secondary internal filter effect.
binding sites.
E-mail address: (W. Minor).
0161-5890/ 2016 Elsevier Ltd. All rights reserved.
K.B. Handing et al. / Molecular Immunology 71 (2016) 143–151
in the drop approximately 10 mM. Crystals were flash-cooled using
phenylmethyl]piperazin-1-yl]ethoxy]acetic acid), which is sold
mineral oil as a cryoprotectant.
under the trade names Zirtec, Zyrtec, and Reactine, is a potent his-
tamine H1 receptor antagonist that is used to treat seasonal allergic
2.2.2. Data collection, structure determination, refinement, and
rhinitis, perennial allergies, and idiopathic urticaria
Cetirizine was the first marketed drug from the
Diffraction data for ESA in complex with cetirizine were col-
series of second-generation antihistamines showing both minimal
lected at a wavelength corresponding to the selenium absorption
side effects on the central nervous system and a reduced level
edge (12670 eV), from a single crystal at 100 K, at the LS-CAT 21-
of cardiotoxicity. Cetirizine exists physiologically as a zwitterion,
ID-D beamline at the Advanced Photon Source, Argonne National
and differs significantly in molecular structure compared to the
Laboratory (Argonne, IL). The data were integrated and scaled by
first-generation H1 receptor antagonists (It is
a racemic mixture of two enantiomers, levocetirizine (R-cetirizine)
structure was solved by molecular replacement using the native
and dextrocetirizine (S-cetirizine), the former being more potent
structure of the ESA protein (PDB ID: 3V08) as the template model.
At therapeutic concentrations in the blood,
Initial electron density maps and model were obtained with HKL-
approximately 90% of cetirizine binds to plasma proteins, primar-
3000 which integrates MOLREP
ily to SA There were multiple attempts to
and auxiliary programs from the CCP4 package
predict cetirizine binding sites on SA, though the sites predicted
Several interactive cycles of model rebuild-
were inconsistent between the studies (
ing and refinement were carried out by HKL-3000 interacting with
Specifically, the
reports variously predicted that cetirizine bound to either drug
Atomic displacement parameters were modeled using indi-
site 1, drug site 2, or both.
vidual isotropic B-factors with translation/libration/screw (TLS)
In this study we present the first crystal structure of a mam-
parametrization to describe anisotropic displacement of those
malian SA, equine serum albumin (ESA, common horse), in complex
atoms. Seven TLS groups were introduced as suggested by the
with cetirizine. In this structure, cetirizine is bound in two sites
TLSMD server Standalone version of
that we label as CBS1 and CBS2. We determine the constants for
MolProbity and the PDB validation tools
the binding of cetirizine to CBS2 in both ESA and human serum
were used for structure quality assessment. Dataset
albumin (HSA) using fluorescence tryptophan quenching (TFQ). We
parameters and structure refinement statistics are summarized
also compare cetirizine binding by ESA and HSA by examining the
in The atomic coordinates and structure factors were
structural conservation of the binding sites. We hypothesize how
deposited in the PDB with identifier 5DQF. The diffraction images
cetirizine binding is affected by the presence of fatty acids and
are available on Integrated Resource for Reproducibility in Macro-
discuss the potential consequences of SA glycation on cetirizine
molecular Crystallography website
transport. In addition, we present an overview of SA drug binding
capacity based on all SA crystal structures available in PDB.
2.3. Tryptophan quenching assay
2. Material and methods
2.3.1. Sample preparation
Both ESA and HSA were dissolved in phosphate-buffered saline
(PBS) buffer (137 mM NaCl, 2.7 mM KCl, 90 mM Na2HPO4, and
pH 7.4) and subsequently purified by size exclu-
Cetirizine dihydrochloride was purchased from Sigma-Aldrich
sion chromatography using a Superdex 200 column attached to
(St. Louis, MO, LOT# 042M4707V, Catalog# C3618). ESA was
an ÅKTA FPLC gel filtration system (GE Healthcare) at 4 ◦C. The
purchased from Equitech-Bio (Kerrville, TX, LOT# ESA62-985,
fractions corresponding to monomeric protein were combined and
Catalog# ESA62) and HSA from Sigma-Aldrich (LOT# SLBD7204,
concentrated. A portion of the purified ESA was dialyzed overnight
Catalog# A3782), both as defatted lyophilized powder. After
into two additional buffers: 100 mM HEPES with 150 mM NaCl at
dissolving the albumin proteins in aqueous solutions, the con-
pH 7.4, and 100 mM Tris with 150 mM NaCl at pH 7.4, and used to
centrations were calculated spectrophotometrically by measuring
test the influence of the buffers on cetirizine binding.
the absorbance at 280 nm (assuming
280-HSA = 28730 M−1 cm−1,
Cetirizine was dissolved in the same buffer as the protein to
280-ESA = 27400 M−1 cm−1, MWHSA = 66470 Da, MWESA = 65700 Da,
two final concentrations of 10 and 7.5 mM respectively, and the
and path length = 1 cm) measured with a Shimadzu UV-2450 UV
pH of each was adjusted to 7.4 in order to be close as possible
spectrophotometer (Kyoto, Japan).
to physiological conditions. Each of the two cetirizine stocks was
subsequently diluted 1:1 with the buffer sequentially 14 times,
2.2. Structure determination
giving a series of 30 dilutions all used for the TFQ assay. Prior to
the TFQ assay, the protein and cetirizine solutions were filtered
2.2.1. Protein purification and crystallization
using 0.1 m membrane filter (Millipore, Billerica, MA, Catalog#
ESA was dissolved in 10 mM Tris pH 7.5 and 150 mM NaCl buffer,
UFC30VV00), degassed, and mixed in a 1:1 ratio.
and was further purified using a Superdex 200 column attached to
an ÅKTA FPLC gel filtration system (GE Healthcare) at 21 ◦C. Fol-
2.3.2. Model used to determine cetirizine dissociation constant by
lowing gel filtration, fractions containing monomeric protein with
a molecular weight of 55–60 kDa were combined and concentrated
Since the total concentration of albumin used was significantly
to 30 mg/mL. Crystals of native ESA were obtained by hanging-
lower than the total concentration of cetirizine (i.e. [P] « [L]), we
drop vapor diffusion on 24-well plates (Qiagen). Plates were set
used a simplified model of TFQ that assumes that the concentration
up manually and observed under the microscope. Well-diffracting
of free ligand is approximated by the concentration of added ligand
crystals grew after 1 day with the reservoir solution composed of:
100 mM Tris HCl pH 7.5, 1800 mM (NH4)2SO4, 87.5 mM NaBr, 2.5%
w/v PEG 8 K. Crystals were soaked with a 100 mM stock of cetirizine
in 100 mM Tris buffer pH 7.4 with a final concentration of cetirizine
K.B. Handing et al. / Molecular Immunology 71 (2016) 143–151
protein absorbance at 280 nm and A
ligand absorbance at
Crystallization data and refinement statistic of ESA in complex with cetirizine.
ESA in complex with cetirizine
2.3.3. Tryptophan quenching measurements
Tryptophan fluorescence intensity was measured at 37 ◦C on
Unit-cell parameters (Å, ◦)
a = 94.2, b = 94.2, c = 141.9
a Pherastar FS (BMG Labtech, Offenburg, Germany) device using
␣ =  = 90, ␥ = 120
two filters: 280 nm for the excitation beam, and 340 nm for the
80.00 − 2.15 (2.19 −
fluorescence detection. Sample solutions with a volume of 100 L
were placed on UV-transparent, half-area 96-well plates (Corning®,
Total number of reflections
One Riverfront Plaza, NY, Catalog# CLS3635). The albumin con-
No. of reflections (refinement/R
centration of 6.1 M for both ESA and HSA was chosen based on
CC ½−highest resolution shell
preliminary experiments. The cetirizine concentration varied from
0.23 M to 5 mM. The gain value (amplification used to increase
the observable signal) was adjusted to 645 and 666 for ESA and
Structure refinement
HSA respectively. The focal height was set to 6.7 mm to ensure the
best signal-to-noise ratio.
Bond lengths RMSD (Å)
The albumin fluorescence intensity for each cetirizine concen-
Bond angles RMSD (◦)
tration was averaged based on three independent experimental
Mean B value (Å2)
repetitions (each repetition was obtained by measuring the fluo-
Number of protein atoms
Mean B value for protein atoms (Å2)
rescence of the well 10 times and averaging the acquired values).
Number of water molecules
The averaged values of three independent measurements of cet-
Mean B value for water molecules (Å2)
irizine fluorescence in control solutions (without protein) were
Number of ligand/ion atoms
subtracted as a background correction for each cetirizine concen-
Mean B value for ligand/ion atoms (Å2)
tration. Experimental errors were estimated as standard deviations
Clashscore percentile (%)
of the obtained values. Values of absorbance for the calculation
Rotamer outliers (%)
of pIFE correction were measured from the same samples. Ap
Ramachandran outliers (%)
was measured from the well with zero cetirizine concentration
Ramachandran favored (%)
recorded for each cetirizine concentration in the
Residues with bad bonds (%)
presence of protein. Afterwards correction factor G (3) for each cet-
Residues with bad angles (%)
irizine concentration was calculated and the values of fluorescence
intensity were multiplied by the correction factor. The TFQ model
a Values in parentheses are for the highest resolution shell.
was fit by nonlinear regression method to the corrected experi-
Ii (hkl) − I (hkl) /
Ii (hkl) , where (I(hkl)) is the mean
mental data with the Origin 6 software (OriginLab Corporation).
of I observations Ii(hkl) of reflection hkl.
c RworkandRfree =
Fo , where Fo and Fc are the observed and
2.4. Sequence and structure analysis
calculated structure factors, respectively, calculated for working set (R
5% of the data, which were omitted from refinement (Rfree).
PyMOL (Schroedinger LLC) was used to superpose the structures
and generate figures. Root-mean-square deviation (RMSD) val-
F is the measured fluorescence, F0 is the fluorescence of the protein
ues were calculated using PyMOL's align function with the cycles
without ligand, Fc is the fluorescence of fully complexed protein, K
parameter set on zero (command: align mobile structure, tar-
is the binding constant and [L] is the ligand concentration. When
get structure, cycles = 0, transform = 0). PyMOL's script color h.py
equation 1 is solved for F, where f = F0 − Fc/F0 is the efficiency of
was used to color-code structure by hydrophobicity according to
quenching, we get:
the Eisenberg hydrophobicity scale BioEdit
(Ibis Biosciences) was used for sequence manipulation and repre-
sentation. The Mafft multiple-sequence alignment server was used
to align the sequences (In the text, the
However, this model does not correct for the so-called "internal
numbering of residues always refers to the HSA sequence number-
filter effect" (IFE) Cetirizine does
ing for simplicity.
not absorb significantly at the tryptophan emission wavelength of
340 nm (secondary IFE or sIFE) and any correction concerning the
absorbance of the fluorescent signal was unnecessary (Supplemen-
2.5. Analysis of ligand binding sites on SA
tary Fig. 1). However, the absorbance at the excitation wavelength
280 nm (primary IFE or pIFE) is significant, especially at higher ceti-
The albumin structures used for the analysis of binding sites
rizine concentrations, and a correction had to be applied to changes
were found using three approaches. First, Dali server (
in fluorescence yield upon increase in ligand concentration.
was run using the structure of HSA (PDB ID:
We used the correction proposed by
1E78) to find structures with a Z-value higher than 9 (below this
for experimental setups where all points of the sample well are
score no molecule was annotated as SA). The resulting list of 142
read by the detector at a constant angle. This condition is true for
chains was further reduced to 132, by excluding protein chains with
the plate-reader with front (top) face geometry which was used
folds different than that of albumin. Second, the PDB tool was used
in the experiment. This correction compensates for fluorescence
to search for structures with sequence similarity 30% and higher
decrease due to pIFE by multiplying the measured fluorescence by
to that of HSA. The search resulted in a set of 103 structures (147
a correction factor G, defined as:
chains). Third, the PDB was searched for all records with the text
‘serum albumin' in the molecule name field. This search produced
106 structures (149 chains). The union of all three approaches
1 − e−(Ap+Al)
resulted in a set of 151 unique chains. Only chains with bound lig-
K.B. Handing et al. / Molecular Immunology 71 (2016) 143–151
ands with more than 5 non-hydrogen atoms were superposed and
Cetirizine's carboxylate group projects outside the pocket and is
further analyzed (93 chains).
bound by an electrostatic interaction with Arg209.
The C␣ atoms superposition of native ESA (PDB ID: 3V08) on the
structure of ESA in complex with cetirizine shows a RMSD value
3. Results
of 1.27 Å (over 580 atoms). The superposition of all atoms of all
residues that have at least one atom within 6 Å of the cetirizine
3.1. Overall fold and structure quality
molecule (atoms from native ESA superposed on atoms from ESA
in complex with cetirizine) gave RMSD values of 0.78 Å (78 atoms)
The crystal of ESA in complex with cetirizine adopted space
for CBS1 and 0.48 Å (58 atoms) for CBS2. Therefore, the binding
1 and diffracted to 2.15 Å resolution. The unit cell has
pockets have the same conformation in both structures and there
dimensions of a = 94.2, b = 94.2 and c = 141.9 Å and accommodates
are no significant conformational changes upon cetirizine binding
one ESA monomer per asymmetric unit. The overall fold is the same
in either CBS1 or CBS2.
as in the native structure and maintains the canonical "heart" shape.
The cetirizine compound used for soaking of ESA crystals was
The structure may be divided into three homologous domains: I
a racemic mixture of two enantiomers, dextrocetirizine and levo-
(residues 1–195, from now on residue numbering will refer to HSA),
cetirizine, same as the marketed drug. In both cetirizine binding
II (196–383) and III (384–585), each containing two subdomains (A
pockets the position of the chloride atom is ambiguous and both
and B) composed of 4 and 6 alpha-helices, respectively (
cetirizine enantiomers may be modeled (i.e. the aromatic rings can
The refined model has good overall geometry with a very low
be swapped). Judging by the best fit to the 2mF
percentage of rotamer outliers and a low MolProbity clash score
chloride atom B-factor, and Real Space R-factor (RSR) value we
The structure has no Ramachandran plot outliers. Accord-
modeled only predominant conformation in each of these pockets:
ing to the amino acid sequence, the mature protein consists of
dextrocetirizine in CBS1 and levocetirizine in CBS2. The cetirizine
582 resides (NCBI accession code: NP 001075972), though three
molecule in CBS1 was refined with an overall occupancy of 1 and
N-terminal residues are disordered and therefore are not visi-
an average B-factor of 78 Å2, and in CBS2 with an occupancy of 0.8
ble in the electron density map. Interestingly, in contrast with
and an average B-factor of 81 Å2. The two cetirizine molecules fit
the previously deposited in PDB structures of Equus caballus SA
well to the electron density map (is supported by low
(PDB IDs: 3V08, 4J2V, 4OT2, 4F5U, and 4F5T), a single point muta-
RSR (0.16 and 0.20 respectively) and Local Ligand Density Fit (LLDF;
tion, R561A, is observed. The long arginine side chain cannot be
1.79 and 2.04 respectively) values.
modeled in this position due to steric clashes with the nearby
disulfide bond connecting Cys567 and Cys558 and a symmetry-
3.3. Conservation of CBS1 and CBS2 between ESA and HSA
related copy of the molecule. Moreover, there is no 2mF
map supporting placement of the side chain. According to the NCBI
Mature mammalian SA proteins have very high sequence con-
database, this mutation is characteristic for Equus ferus przewalskii,
servation across different species. HSA is 84.8% similar and 76.2%
a rare subspecies of wild horse from central Asia (accession code:
identical by sequence as compared to ESA. This high sequence con-
XP 008524663.1). However it is possible that there is an error in
servation is reflected in the conserved three-dimensional structure,
the Equus caballus SA sequence, or the observed mutation naturally
and allows the protein to maintain the same functions in different
occurs in that species.
species Since there are no major confor-
mational changes in ESA upon binding of cetirizine, the binding
3.2. Cetirizine binding sites in equine serum albumin
sites in ESA may be directly compared to corresponding sites in
the apo-structure of HSA (PDB ID: 1E78), after overall C␣ atoms
Defatted ESA was used for crystallization in order to avoid
superposition of the two structures. The comparison shows that
potential competition of cetirizine and fatty acids for the same
the geometries of binding sites CBS1 and CBS2 are well maintained
binding site. Two cetirizine molecules per ESA monomer were
found in the crystal structure The first cetirizine
CBS1 in ESA is composed of 16 residues, defined as those located
binding site (CBS1) is situated in a deep pocket located at the inter-
within 6 Å of the cetirizine molecule with side chains that are facing
face between subdomains IA and IB, where cetirizine is "wedged"
the pocket (10 (62.5%) of these residues are fully conserved
in a crevice between helices h2 and h3 from subdomain IA, and
in HSA and five (31.3%) are similar (The only significant
h2 from IB. This site is hydrophobic due to the presence of seven
change in CBS1 is Lys136 of HSA, which corresponds to a glycine
highly hydrophobic resides: Leu24, Phe36, Val40, Val43, Leu135,
residue in ESA. Lys136 is buried inside the pocket and may poten-
Leu139, and Leu155 (The two aromatic rings of the cetirizine
tially interfere with cetirizine binding in HSA, especially taking into
molecule are locked in the binding pocket due to hydrophobic inter-
account that this residue may be glycated in humans (see Section
actions. Additionally, the OD1 and OD2 atoms of Asp132, which
However, when comparing the structure of HSA superposed on
are located at the opening of the binding cavity, form a strong
the structure of ESA in complex with cetirizine, Lys136 does not
salt bridge to the NAF nitrogen atom of the piperazine ring and
form clashes with cetirizine molecule. Moreover, Lys136 is located
further stabilize cetirizine in this position. The carboxylate group
at the entrance to CBS1 and demonstrates high flexibility; therefore
of cetirizine protrudes outside the pocket and interacts with a
the side chain may easily flip away from the ligand, leaving space
symmetry-related copy of the protein. The oxygen atom OAC of
for cetirizine to bind. The Asp132 residue forming the salt bridge
the carboxylate group of cetirizine forms a salt bridge with nitro-
with the piperazine ring of cetirizine molecule in CBS1 corresponds
gen atom NE2 from His338 with a symmetry-related copy of the
to Glu in HSA, which should preserve the electrostatic interaction
protein, which locks the tail in a defined conformation.
given the conformational flexibility of the residue.
Cetirizine binding site 2 (CBS2) is located in a pocket formed by
CBS2 in ESA is composed of 13 residues, defined in the same
helix h2 from subdomain IIA and h2 and h3 from subdomain IIB.
manner as for CBS1. 12 (92.3%) of them are fully conserved in HSA
Similarly to CBS1, the two aromatic rings of the cetirizine molecule
The only difference in HSA is Val482, which corresponds to
create hydrophobic interactions with five highly hydrophobic
alanine in ESA. This substitution should not create any clashes with
resides which coat the pocket: Val216, Phe228, Leu327, Leu331
the cetirizine molecule and may even slightly increase the propen-
and Leu347 The piperazine ring is stabilized by a strong
sity of CBS2 to bind cetirizine, since valine is more hydrophobic than
salt bridge between the NAF atom and OE1 and OE2 from Glu354.
alanine (In addition, the residue forming the salt bridge with
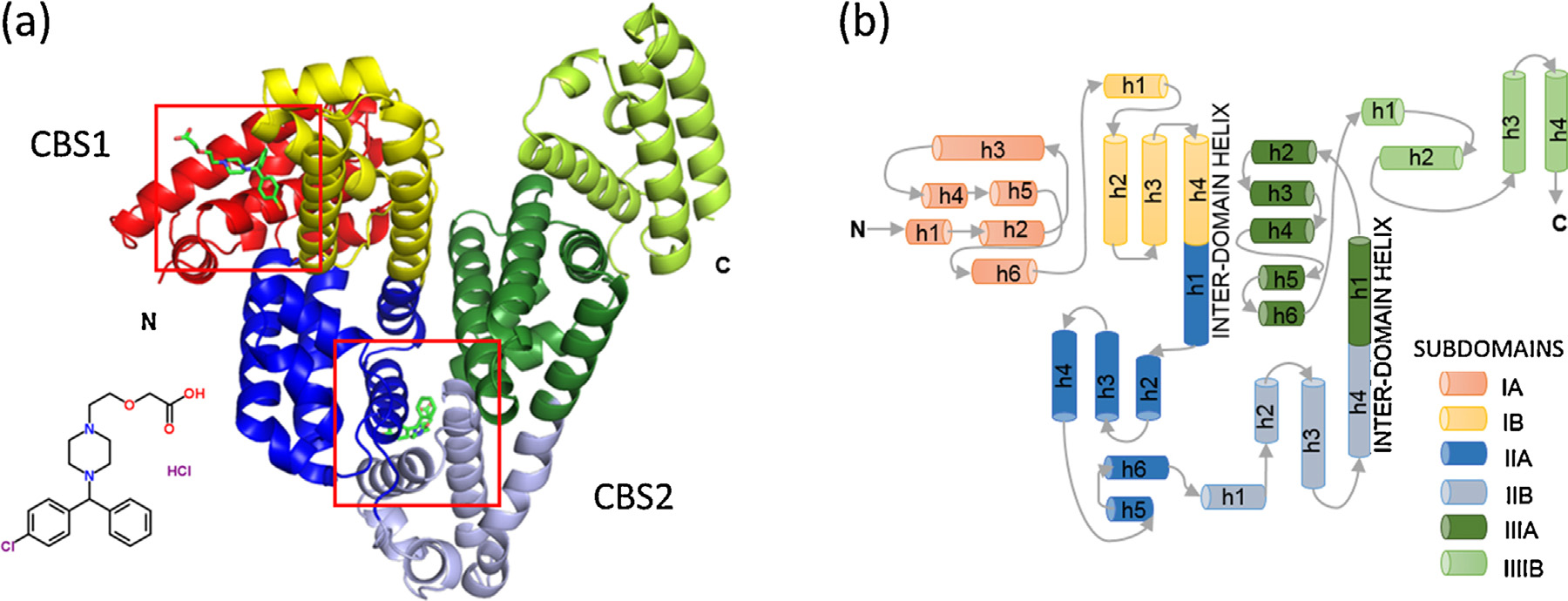

K.B. Handing et al. / Molecular Immunology 71 (2016) 143–151
Fig. 1. (a) The overall fold of equine serum albumin structure in complex with cetirizine. Helices are represented by ribbons, and cetirizine molecules are shown in green
in stick representation and marked with red squares. (b) Topology diagram of albumin. Helices are represented by cylinders, and loops by arrows. Subdomains are colored
according to the legend. Names of helices take the form ‘h1�, where ‘h' stands for helix and numbers indicate the order of the helices in each subdomain. (For interpretation
of the references to color in this figure legend, the reader is referred to the web version of this article.)
binding of cetirizine to CBS2, because the tryptophan residue is too
far away from CBS1 for its fluorescence to be disrupted by cetirizine
binding in CBS1.
After applying correction for pIFE, the obtained fluorescence
data fits well into the model described by the equation 1
However, the observed decrease in the intensity of fluorescence
may be the result of collisional quenching. In the case of collisional
quenching, the bimolecular quenching rate constant kq cannot be
larger than 2 × 1010 M−1 s−1 The
kq calculated from obtained K
rules out this possibility:
is the fluorescence lifetime of the fluorophore without ligand;
in the case of tryptophan this value is in the range of 1–10 ns.
Accounting for pIFE and ruling out collisional quenching leaves
direct binding of the ligand as the most likely cause of decrease
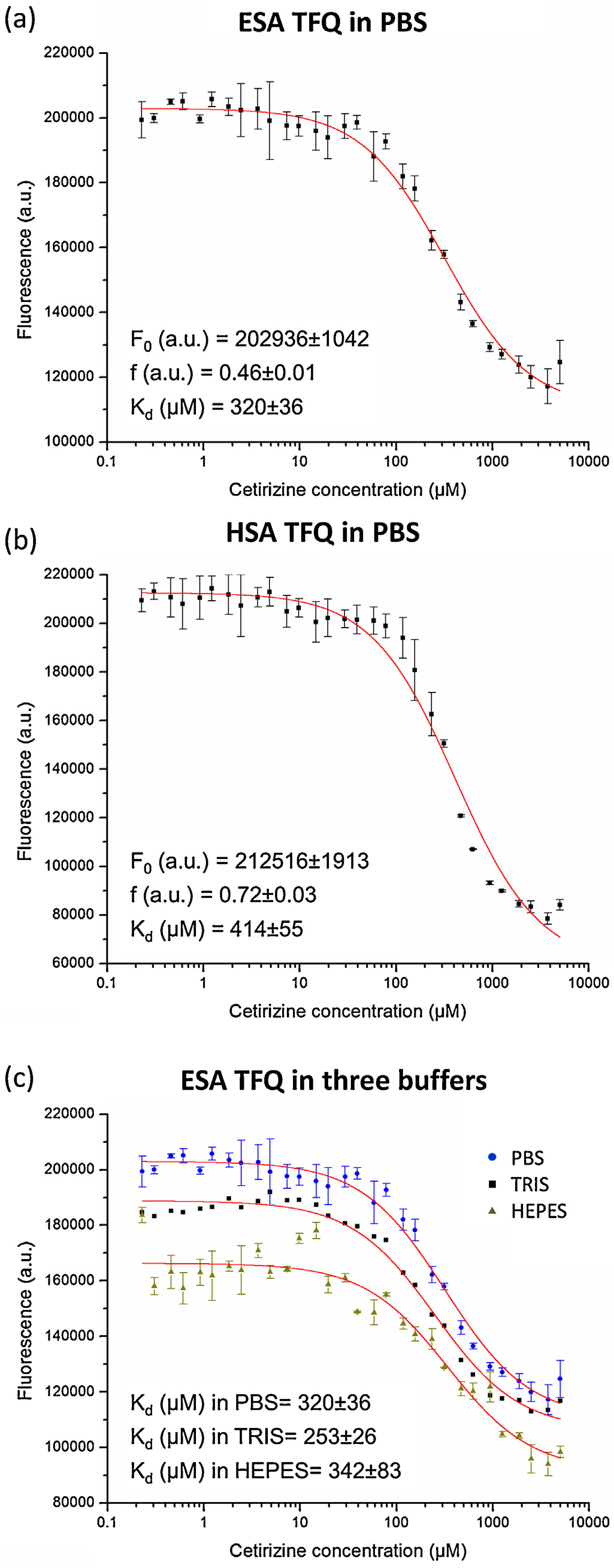
in fluorescence intensity.PBS buffer at pH 7.4 was selected for TFQ
experiments for both albumins, since it best mimics the native
blood conditions. The calculated dissociation constants K
irizine in CBS2 are 320 ± 36 M for ESA and 414 ± 55 M for HSA.
We also analyzed the influence of three commonly used buffers –
Tris, HEPES, and PBS, all at pH 7.4 – on cetirizine binding to ESA. As
Fig. 2. Cetirizine binding sites in ESA. (a, b) CBS1, (c, d) CBS2. Cetirizine molecules
shown on buffers do not have a significant influence on
are shown in sticks, carbon marked in green, chloride in orange, nitrogen in blue,
cetirizine binding to ESA as measured by TFQ.
and oxygen in red. Helices are marked by the subdomain name and helix number
as proposed in (a, c) Cetirizine shown in its binding environ-
ment. The 2mFo-DFc omit electron density maps are presented in orange ( − 0.8).
3.5. Comparison of CBS1 and CBS2 with other drug binding sites
(b, d) The protein surface in the cetirizine binding environment is color-coded by
hydrophobicity. Color scales from red to white, where red indicates more hydropho-
bic and white more hydrophilic. (For interpretation of the references to color in this
figure legend, the reader is referred to the web version of this article.)
Contrary to the previous non-crystallographic studies, none of
the cetirizine molecules are located in the main drug site 1 or in
drug site 2 (CBS1 does not
the piperazine ring in CBS2 (Glu354) is also conserved, suggesting
overlap with any of the three main drug binding sites, nor the fatty
that the electrostatic interaction is conserved in cetirizine binding
acid binding sites, suggesting that CBS1 is a new drug binding site,
to HSA as well.
labeled as site 4 on A superposition of all available in PDB
SA structures with small molecules bound showed only one struc-
3.4. Cetirizine dissociation constant at CBS2 in ESA and HSA
ture with a drug bound in CBS1 (PDB ID: 1E7C). In the publication
describing 1E7C, the interaction of halothane (general anesthetic
Both ESA and HSA have only one tryptophan residue in the entire
drug) in this position was concluded by the authors to be a crys-
amino acid sequence. It is located at position Trp214, which is about
tallographic artifact, as it occurs only in the presence of fatty acids
33 Å from CBS1 and about 7.8 Å from CBS2. Since TFQ occurs only if
and the binding was forced by the close interaction of an asym-
the distance between the quencher and tryptophan residue is short
metric copy of the protein in the crystal structure (
(∼5–15 Å) (TFQ is only suitable for monitoring
Four other small molecule ligands were found to be
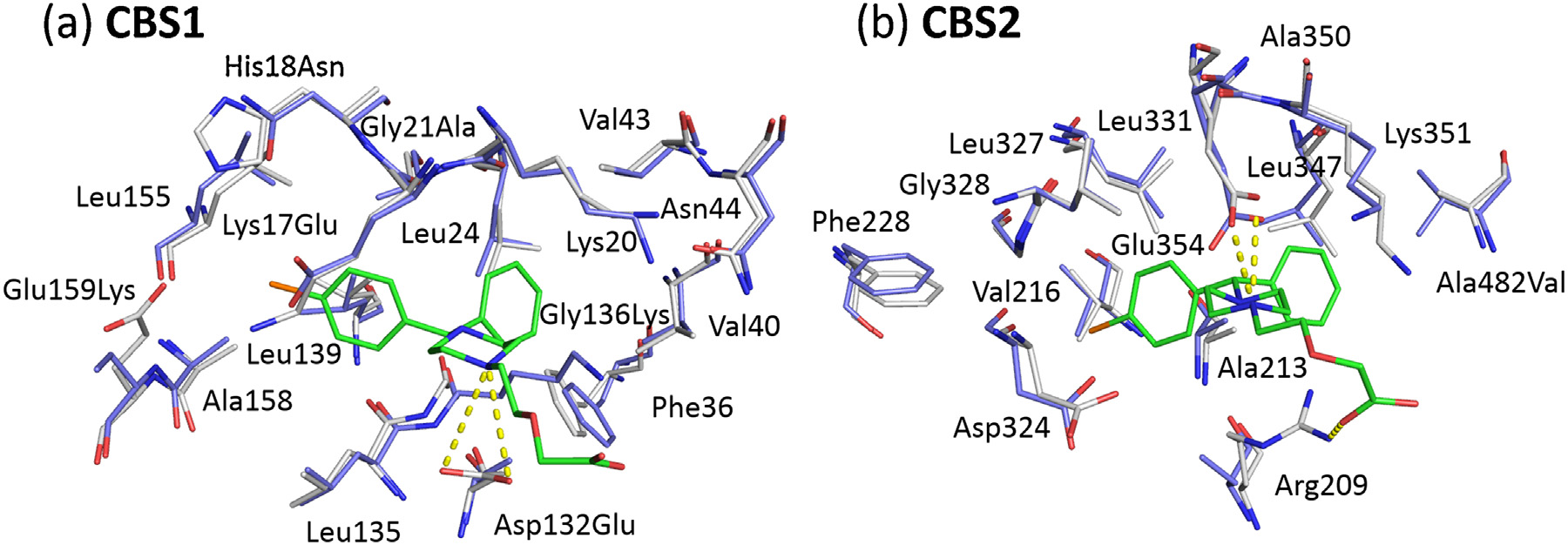

K.B. Handing et al. / Molecular Immunology 71 (2016) 143–151
Fig. 3. Superposition of cetirizine binding residues from ESA (white, PDB ID: 5DQF) and HSA (blue, PDB ID: 1E78) in CBS1 (a) and CBS2 (b). Cetirizine molecules are shown in
sticks, carbon marked in green, chloride in orange, nitrogen in blue, and oxygen in red. Resides that are not conserved between ESA and HSA are labeled with the ESA residue
name followed by the HSA residue number and the HSA residue name. The salt bridges between cetirizine and ESA are depicted with dashed yellow lines. (For interpretation
of the references to color in this figure legend, the reader is referred to the web version of this article.)
Fig. 4. Sequence alignment of HSA and ESA. Residues forming the CBS1 in ESA are marked in green, and CBS2 in red. Residues that are conserved in HSA are highlighted in
a darker shade and similar residues are in a lighter shade. Among them, highly hydrophobic residues are marked with rectangles. The arrows indicate residues involved in
cetirizine binding and shown to undergo glycation in certain conditions (such as diabetes). (For interpretation of the references to color in this figure legend, the reader is
referred to the web version of this article.)
bound in CBS1: acetate ion, malonate ion, polypropylene glycol, and
zone, the anti-inflammatory drug naproxen, and 3,5-diiodosolicylic
acid. Other small molecules found in this pocket include the radio-
CBS2 overlaps with previously described fatty acid binding site
contrast agent iophenoxic acid, and three molecules originating
FA6 (Other drugs that were found in CBS2 are: halothane,
from crystallization screens: polyethylene glycol, triethylene gly-
ibuprofen, and 3,5-diiodosolicylic acid (a medication for the treat-
col, and l-malic acid.
ment of parasitic intestinal worms). Moreover, the following
crystallization agents were found in this pocket: polyethylene gly-
col, formic acid, and malonate ion.
The superposition of all available in PDB structures of SA showed
Herein we investigated the interaction between the antihis-
two other binding sites not corresponding to either CBS1 or the
tamine drug cetirizine and ESA using X-ray crystallography and TFQ
previously described main drug or fatty acid binding sites, labeled
techniques. The crystal structure revealed two cetirizine binding
as site 5 and 6 on Site 5 is characterized by a structure
sites on ESA: one site that constitutes a novel drug binding pocket
with the anti-cancer drug etoposide bound (PDB ID: 4LB9) and is
(CBS1 or drug site 4), and the second site coincides with the known
located between subdomains IB and IIA. There is no other ligand
fatty acid binding site 6 (CBS2 or FA6). The binding is ensured by
present in this position in any other SA structure, likely due to the
highly hydrophobic pockets, with hydrophilic residues additionally
fact that the pocket spans on the interface of three flexible sub-
stabilizing cetirizine at the entrance to the binding cavities via for-
domains: IA, IIA, and IB. Site 6 is located on the interface between
mation of salt bridges. Analysis of the 2mF
subdomains IIIA and IIIB. There are three drugs bound in this site
vicinity of the cetirizine binding sites suggests that both cetirizine
in various SA structures: the anti-inflammatory drug oxyphenbuta-
enantiomers may bind in both CBS1 and CBS2, but only the pre-
K.B. Handing et al. / Molecular Immunology 71 (2016) 143–151
dominant molecule, dextrocetirizine in CBS1 and levocetirizine in
CBS2, was modeled in the final structure.
The lack of a crystal structure of HSA in complex with cetirizine
hindered direct comparison of cetirizine interaction with ESA in
relation to HSA. However, structural comparison of ESA and HSA
shows that the hydrophobic environment and salt bridges for both
CBS1 and CBS2 are preserved in HSA due to high sequence conser-
vation, with CBS2 in particular being virtually identical to the site
in ESA. Consequently, we can expect the same mode of cetirizine
binding to HSA as observed in ESA.
This conclusion is further strengthened by the results of TFQ
experiment measuring cetirizine binding in CBS2 for ESA and
HSA. The dissociation constants are identical within experimental
error and are in the agreement with previous reports measuring
the binding of cetirizine to HSA.
using equilibrium dialysis two cetirizine binding sites on HSA: K
weak = 200 M, and K
single cetirizine binding site on HSA with K
776 M at 318 K by
TFQ. Using the same method, that
cetirizine binds to a single site on HSA and BSA with K
373 M at 308 K, respectively.
Despite the significant structural conservation of CBS1 and CBS2,
and similarity in the CBS2 binding constants detected by TFQ, the
binding sites in the ESA crystal structure differ from those that have
been previously proposed for HSA. Equilibrium dialysis of cetirizine
binding to HSA in the presence of site markers warfarin (drug site
1) and diazepam (drug site 2) indicated that both sites are involved
in cetirizine binding
concluded that cetirizine binds with higher affinity to drug site 2
and lower affinity to drug site 1. Bree et al. showed that levoce-
tirizine binds to three different binding sites on HSA: one strong
site overlapping with drug site 2 and two weak sites with very low,
non-specific affinity. In the same study, the binding of levocetirizine
to HSA was also shown to decrease in the presence of fatty acids,
which was concluded to be consistent with levocetirizine binding to
drug site 2 (An independent study of cetirizine and
levocetirizine binding to HSA using TFQ detected just one binding
site in which levocetirizine was shown to compete with diazepam
(drug site 2) for binding (Conversely, a recent study
of cetirizine binding to BSA and HSA using TFQ found that cetirizine
bound primarily at drug site 1
These discrepancies have two feasible explanations: (a) the
actual sites for cetirizine binding on SA revealed by the crystal struc-
ture of ESA could not be resolved using other methods, or (b) HSA
has different binding sites for cetirizine than ESA despite the level
of sequence and structure conservation and similarity of binding
constants. Differences in binding sites for HSA versus SA from other
mammals have been reported for other ligands (
This raises the issue of potential differences in drug transport and
delivery between human and animal models used in clinical stud-
ies, and highlights the importance of structural studies of albumins
from species other than human.
Nonenzymatic glycosylation (NEG or glycation) results in the
addition of reducing sugars and/or their reactive degradation prod-
ucts to proteins in blood plasma (This
process is promoted by the presence of elevated blood glucose con-
centrations in patients with diabetes, and affects various proteins
including HSA (In certain diabetic states, the
binding of drugs to HSA can be significantly altered
In addition, levocetirizine was indicated to bind
more strongly to glycated HSA than to non-glycated HSA
Fig. 5. TFQ in ESA and HSA by cetirizine. Graphs show tryptophan fluorescence
An analysis of lysine and arginine residues in the context
intensity after applying correction for pIFE vs. cetirizine concentration. Error bars
of glycation indicates that several residues in CBS1 and CBS2 may
represent standard deviation of fluorescence intensities from three independent
potentially be glycated. There are two such residues in ESA fac-
replicates of the experiment. TFQ in ESA (a) and HSA (b) as a function of cetirizine
concentration in PBS buffer. (c) Influence of three different buffers on cetirizine
ing the ligand in the CBS1 pocket, Lys17 and Lys20. Moreover, HSA
binding in ESA: HEPES, Tris, and PBS. F
has one additional lysine sticking into the pocket, Lys136. Among
0 –fluorescence of protein without ligand,
Kd—binding constant in M, and f—the efficiency of quenching.
K.B. Handing et al. / Molecular Immunology 71 (2016) 143–151
Fig. 6. Summary of the ligand binding capacity of SA based on all crystallographic structures of SA currently deposited in PDB. Helices are shown in ribbon representation,
and ligand atoms in spheres. The name of each binding site is labeled, CBS in red, drug binding sites that do not overlap with known FA binding sites in blue. All of the atoms
of all ligands found in a particular site are colored in one color. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version
of this article.)
these residues, Lys20 and Lys136 have been shown to be glycated
Tri-Corridor (Grant 085P1000817). The work described here was
in HSA in vitro In the CBS2, there are two
supported by NIH grants GM094662, GM053163, HG008424.
residues, Arg209 and Lys351, that could be potentially glycated,
and both were reported to undergo glycation in HSA (
Appendix A. Supplementary data
We conclude that for patients
with diabetes the affinity of cetirizine to SA can be affected by SA
Supplementary data associated with this article can be found,
glycation. Although the exact effect of SA glycation on cetirizine
in the online version, at
binding is still unknown, it may be necessary to adjust the amount
of administrated drug for patients with diabetes in order to achieve
the same therapeutic effects.
Our review of all crystal structures of SA in complex with small
molecules available in the PDB shows that in addition to the novel
Anguizola, J., Matsuda, R., Barnaby, O.S., Hoy, K.S., Wa, C., DeBolt, E., Koke, M., Hage,
drug binding site CBS1, there are two other drug binding sites
D.S., 2013. Review: glycation of human serum albumin. Clin. Chim. Acta 425,
drug sites 5, 6) that do not overlap with fatty acid binding sites. Con-
sequently, the drugs bound in these sites will not compete directly
Barnaby, O.S., Cerny, R.L., Clarke, W., Hage, D.S., 2011. Comparison of modification
sites formed on human serum albumin at various stages of glycation. Clin.
with fatty acids for transport on albumin. On the other hand, the
Chim. Acta 412, 277–285, .
main drug binding sites 1–3 on SA do bind fatty acids, and therefore
Bern, M., Sand, K.M.K., Nilsen, J., Sandlie, I., Andersen, J.T., 2015. The role of albumin
drug binding in these sites (as well as in all other FA binding sites
receptors in regulation of albumin homeostasis: implications for drug delivery.
including CBS2) can be affected by competition with fatty acids.
J. Control. Release 211, 144–162,
Elevated levels of fatty acids (from dietary intake, diabetes melli-
Bhattacharya, A.A., Curry, S., Franks, N.P., 2000a. Binding of the general anesthetics
tus, or myocardial infarction) or decreased levels of albumin (liver
propofol and halothane to human serum albumin: high resolution crystal
disease) may decrease the apparent binding
structures. J. Biol. Chem. 275, 38731–38738,
of drugs in these sites.
Bhattacharya, A.A., Grüne, T., Curry, S., 2000b. Crystallographic analysis reveals
The results presented here describe the interaction between SA
common modes of binding of medium and long-chain fatty acids to human
and the antihistamine drug cetirizine on the molecular level, con-
serum albumin. J. Mol. Biol. 303, 721–732,
tribute to further understanding of drug transport and delivery in
Bree, F., Thiault, L., Gautiers, G., Benedetti, M.S., Baltes, E., Rihoux, J.P., Tillement,
mammals, and potentially will inform further rational drug design.
J.P., 2002. Blood distribution of levocetirizine, a new non-sedating histamine
H1-receptor antagonist, in humans. Fundam. Clin. Pharmacol. 16, 471–478,
Chen, V.B., Arendall, W.B., Headd, J.J., Keedy, D.A., Immormino, R.M., Kapral, G.J.,
Murray, L.W., Richardson, J.S., Richardson, D.C., 2010. MolProbity: all-atom
structure validation for macromolecular crystallography. Acta Crystallogr. D.
Biol. Crystallogr. 66, 12–21,
We would like to thank Matthew D. Zimmerman, Jonathan
Chruszcz, M., Mikolajczak, K.B., Mank, N., Majorek, K.A., Porebski, P.J., Minor, W.,
2013. Serum albumins—unusual allergens. Biochim. Biophys. Acta Gen. Subj.
Handing and David Cooper for critical reading and discussion. We
1830, 5375–5381, .
also thank beamline scientists for assistance in data collection. The
Curry, S., 2009. Lessons from the crystallographic analysis of small molecule
structural data shown in this report used resources of the Advanced
binding to human serum albumin. Drug Metab. Pharmacokinet. 24, 342–357,
Photon Source, a U.S. Department of Energy (DOE) Office of Science
Ehrenberg, M., Cronvall, E., Rigler, R., 1971. Fluorescence of proteins interacting
User Facility operated for the DOE Office of Science by Argonne
with nucleic acids. Correction for light absorption. FEBS Lett. 18, 199–203,
National Laboratory under Contract No. DE-AC02-06CH11357. Use
Eisenberg, D., Schwarz, E., Komarony, M., Wall, R., 1984. Analysis of membrane and
of the LS-CAT Sector 21 was supported by the Michigan Eco-
surface protein sequences with the hydrophobic moment plot. J. Mol. Biol. 179,
nomic Development Corporation and the Michigan Technology
K.B. Handing et al. / Molecular Immunology 71 (2016) 143–151
Emsley, P., Cowtan, K., 2004. Coot: model-building tools for molecular graphics.
Sekula, B., Zielinski, K., Bujacz, A., 2013. Crystallographic studies of the complexes
Acta Crystallogr. D. Biol. Crystallogr. 60, 2126–2132,
of bovine and equine serum albumin with 3,5-diiodosalicylic acid. Int. J. Biol.
Macromol. 60, 316–324, .
Gadgil, H.S., Bondarenko, P.V., Treuheit, M.J., Ren, D., 2007. Screening and
Spector, A.A., John, K., Fletcher, J.E., 1969. Binding of long-chain fatty acids to
sequencing of glycated proteins by neutral loss scan LC/MS/MS method. Anal.
bovine serum albumin. J. Lipid Res. 10, 56–67,
Chem. 79, 5991–5999, .
Ghuman, J., Zunszain, P.A., Petitpas, I., Bhattacharya, A.A., Otagiri, M., Curry, S.,
Sudlow, G., Birkett, D.J., Wade, D.N., 1975.
2005. Structural basis of the drug-binding specificity of human serum albumin.
J. Mol. Biol. 353, 38–52, .
Sudlow, G., Birkett, D.J., Wade, D.N., 1976.
Hegde, A.H., Sandhya, B., Kalanur, S.S., Seetharamappa, J., 2011. Binding mechanism
of bioactive cetirizine hydrochloride to sudlow's site i of serum albumins. J.
Sugio, S., Kashima, A., Mochizuki, S., Noda, M., Kobayashi, K., 1999.
Solut. Chem. 40, 182–197,
Holm, L., Rosenström, P., 2010. Dali server: conservation mapping in 3D. Nucleic
Tillement, J.P., Testa, B., Brée, F., 2003. Compared pharmacological characteristics
Acids Res. 38, 545–549, .
in humans of racemic cetirizine and levocetirizine, two histamine H1-receptor
Katoh, K., Standley, D.M., 2013. MAFFT multiple sequence alignment software
antagonists. Biochem. Pharmacol. 66, 1123–1126,
version 7: improvements in performance and usability. Mol. Biol. Evol. 30,
Vagin, A., Teplyakov, A., 1997. MOLREP: an automated program for molecular
Liu, X., Du, Y., Sun, W., Kou, J., Yu, B., 2009. Study on the interaction of
replacement. J. Appl. Crystallogr. 30, 1022–1025,
levocetirizine dihydrochloride with human serum albumin by molecular
spectroscopy. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 74, 1189–1196,
Van De Weert, M., Stella, L., 2011. Fluorescence quenching and ligand binding: a
critical discussion of a popular methodology. J. Mol. Struct. 998, 145–150,
Mansoor, S.E., DeWitt, M.A., Farrens, D.L., 2012. Distance mapping in proteins using
fluorescence spectroscopy: the tryptophan-induced quenching (TrIQ) method.
Wang, Z., Ho, J.X., Ruble, J.R., Rose, J., Rüker, F., Ellenburg, M., Murphy, R., Click, J.,
Changes 29, 997–1003,
Soistman, E., Wilkerson, L., Carter, D.C., 2013. Structural studies of several
clinically important oncology drugs in complex with human serum albumin.
Minor, W., Cymborowski, M., Otwinowski, Z., Chruszcz, M., 2006. HKL-3000: the
Biochim. Biophys. Acta 1830, 5356–5374,
integration of data reduction and structure solution–from diffraction images to
an initial model in minutes. Acta Crystallogr. D. Biol. Crystallogr. 62, 859–866,
Winn, M.D., Ballard, C.C., Cowtan, K.D., Dodson, E.J., Emsley, P., Evans, P.R., Keegan,
R.M., Krissinel, E.B., Leslie, A.G.W., McCoy, A., McNicholas, S.J., Murshudov, G.N.,
Murshudov, G.N., Skubák, P., Lebedev, A.A., Pannu, N.S., Steiner, R.A., Nicholls, R.A.,
Pannu, N.S., Potterton, E.a, Powell, H.R., Read, R.J., Vagin, A., Wilson, K.S., 2011.
Winn, M.D., Long, F., Vagin, A.A., 2011. REFMAC5 for the refinement of
Overview of the CCP4 suite and current developments. Acta Crystallogr. D. Biol.
macromolecular crystal structures. Acta Crystallogr. D. Biol. Crystallogr. 67,
Crystallogr. 67, 235–242,
Otwinowski, Z., Minor, W., 1997. Processing of X-ray diffraction data collected in
oscillation mode. Methods Enzymol. 276, 307–326,
Pagliara, A., Testa, B., Carrupt, P.A., Jolliet, P., Morin, C., Morin, D., Urien, S.,
R value: Is a measure of the agreement between the crystallographic model and the
Tillement, J.P., Rihoux, J.P., 1998. Molecular properties and pharmacokinetic
experimental X-ray diffraction data
behavior of cetirizine, a zwitterionic H1-receptor antagonist. J. Med. Chem. 41,
Is a measure of how well the current atomic model predicts a subset of
the measured reflection intensities that were not included in the refinement
Painter, J., Merritt, E.A., 2006. TLSMD web server for the generation of multi-group
B factor: Is a measure of how much an atom oscillates or vibrates around the position
TLS models. J. Appl. Crystallogr. 39, 109–111,
specified in the model
RMSD: Is the measure of the average distance between the atoms of superposed
Peters, T.J., 1995.
Space group: Is a designation of the symmetry of the unit cell of a crystal
Read, R.J., Adams, P.D., Arendall, W.B., Brunger, A.T., Emsley, P., Joosten, R.P.,
Kleywegt, G.J., Krissinel, E.B., Lütteke, T., Otwinowski, Z., Perrakis, A.,
Unit cell: Is the most basic and least volume consuming repeating structure of a
Richardson, J.S., Sheffler, W.H., Smith, J.L., Tickle, I.J., Vriend, G., Zwart, P.H.,
2011. A new generation of crystallographic validation tools for the protein data
TFQ: Refers to any process which decreases the fluorescence intensity of a trypto-
bank. Structure 19, 1395–1412, .
Ruiz-Cabello, F., Erill, S., 1984. Abnormal serum protein binding of acidic drugs in
diabetes mellitus. Clin. Pharmacol. Ther. 36, 691–695,
Source: https://repo.pw.edu.pl/docstore/download.seam;jsessionid=4DC4DC7E22FDA5E6D79080A44F7C695B?fileId=WUT8dce11fc09e64896a3e5515995ccc4e3
ENDOMINE – DIABETIC BLOOD TEST RESULTS DATA MINING PROJECT PLAN ENDOMINE DIABETIC TREATMENT KNOWLEDGE EXTRACTION FROM BLOOD TEST DATA OF JGH ENDOCRINOLOGY DEPARTMENT Projet # …. Auteur : Fodil Belghait - ETS Date of issue: Mai 7th 2013
International Journal of Oral & Maxillofacial Pathology. 2012;3(3):56-60 ISSN 2231 – 2250 Available online at http://www.journalgateway.com or www.ijomp.org Case Report Intraosseous Transmigration of Impacted Canines: Report of Five Cases Sulabha AN, Sachin Deshpande, Sameer C Abstract Transmigration is rare and unusual anomaly involving the intraosseous migration of impacted tooth across midline regardless of the distance. This occurs almost exclusively with mandibular canines. Etiology is still unclear and is not well documented in the literature. Transmigration can occur as isolated finding or in association with other findings such as dentigerous cyst, odontomas, impactions of other teeth etc. Transmigrated canines are particularly significant due to aesthetic and functional importance. Early radiographic examination of the patient is important for the treatment. More documentation of this anomaly and future studies may lead to a better understanding of this rare anomaly, its etiology and improvement of the classification criteria. This paper presents five cases of transmigration of mandibular canines of which two were associated with dentigerous cyst, one associated with bodily rotation of canine, one associated with absence of third molars on right side. Keywords: Impacted Canine;Tooth Migration;Transmigration;Intraosseous;Dentigerous Cyst; Observation. Sulabha AN, Sachin Deshpande, Sameer C. Intraosseous Transmigration of Impacted Canines: Report of Five Cases. International Journal of Oral and Maxillofacial Pathology; 2012:3(3):56-60. ©International Journal of Oral and Maxillofacial Pathology. Published by Publishing Division, Celesta Software Private Limited. All Rights Reserved.