Synthesis of isosorbide based polyurethanes: an isocyanate free method


Contents lists available at
Reactive & Functional Polymers
Synthesis of isosorbide based polyurethanes: An isocyanate free method
Vincent Besse , Rémi Auvergne , Stéphane Carlotti , Gilles Boutevin , Belkacem Otazaghine ,Sylvain Caillol , Jean-Pierre Pascault Bernard Boutevin ,
a Institut Charles Gerhardt Montpellier UMR 5253 – Equipe Ingénierie et Architectures Macromoléculaires, Ecole Supérieure de Chimie de Montpellier, 8 rue de l'Ecole Normale,34296 Montpellier Cedex 5, Franceb Specific Polymers, Avenue de l'Europe, 34830 Clapiers, c Laboratoire de Chimie des Polymères Organiques UMR 5629 Université Bordeaux 1/CNRS, Ecole Nationale Supérieure de Chimie, de Biologie & de Physique, 16, AvenuePey-Berland, 33607 Pessac Cedex, Franced Ecole des Mines d'Alès, Centre des Matériaux de Grande Diffusion (CMGD), 6, Avenue de Clavières, 30319 Alès Cedex, Francee University-Lyon, INSA-Lyon, Ingénierie des Matériaux Polymères, IMP, UMR5223, F-69621 Villeurbanne, France
The synthesis of isocyanate free polyurethanes is a major concern. This paper first reports the synthesis of
Received 20 November 2012
new biobased isosorbide dicyclocarbonates from isosorbide. Then polyhydroxyurethanes (PHUs) were
Accepted 4 January 2013
synthesized by a cyclocarbonate–amine step growth polyaddition with four commercial diamines (e.g.
Available online 11 January 2013
jeffamine D-400, 1,10 diaminodecane, diethylenetriamine and isophoronediamine). These unprecedentedproducts, obtained with high yield, were characterized by 1H NMR, FTIR, DSC, SEC and TGA analyses.
PHUs exhibited glass transition temperatures from �8 °C to 59 °C, and degradation temperatures (Td
5%) between 234 °C and 255 °C. Last but not least, the compounds produced during the degradation of
these PHUs were analyzed by ATG-IR technique and showed that carbon dioxide and secondary amines
Renewable resourcesThermal stability
are released.
Ó 2013 Elsevier Ltd. All rights reserved.
Following this route, polyhydroxyurethanes (PHUs) are obtainedwith inter- and intramolecular hydrogen bonds, which are ex-
Generally, linear polyurethanes (PUs) are obtained by the
pected to present higher chemical and hydrolysis resistances.
reaction between an oligomeric diol (low molecular weight poly-
We previously reported several works on the synthesis of new
mer with terminal hydroxyl groups), a short diol as chain exten-
dicyclocarbonates by esterification of diacids such as terephthalic
der and a diisocyanate. To prepare cross-linked PUs, polyols or
acid with glycerol carbonate. These works led to the synthesis of
isocyanates with functionality higher than 2 can be used. How-
PHUs with ester bonds which exhibited poor stability towards
ever the use of diisocyanate should be avoided for several rea-
hydrolysis We overcame this issue with the synthesis of
sons. Isocyanate reactants are generally very harmful for
dicyclocarbonates from allyl-cyclic carbonate and dithiols by
human health, particularly for people exposed during polyure-
thiol–ene coupling . But these dicyclocarbonate were based
thanes synthesis and could entail adverse health effects such
on aliphatic structures. Yet, PUs contain generally rigid segments
as asthma, dermatitis, conjunctivitis and acute poisoning
made of aromatic groups such as toluene diisocyanate (TDI), which
Therefore the synthesis of PUs, from step growth polyaddition
is a hazardous compound. Therefore we intended to synthesize
of dicyclocarbonates and diamines should be favored. This meth-
new biobased PHU with rigid segments and with ether bonds. Iso-
od is particularly interesting since no hazardous isocyanates are
sorbide is obtained from the dehydration of sorbitol, which is a
used. Thus, this old reaction is currently gaining a lot of
product of the sugar industry and contains two cycloaliphatic rings
attention as a substitution route for the synthesis of PUs
likely to provide a good rigidity to the polymer. Its structure is alsocomposed of two secondary hydroxyl groups.
Isosorbide is a biobased platform chemical extensively studied
⇑ Corresponding author. Tel.: +33 467144300; fax: +33 467147220.
in literature with various industrial applications (isosorbide nitrate,
E-mail addresses: (S. Carlotti),
diesters, lubricant and plasticizers, green solvents, etc.) . It
(B. Otazaghine), (B. Boutevin).
has to be noticed that the synthesis of the corresponding isosorbide
1 Tel.: +33 4677144305; fax: +33 467147220.
amine and isocyanate was already reported Applications
2 Tel.: +33 540002734.
3
of isosorbide in polymers and materials are even more important
Tel.: +33 466785669.
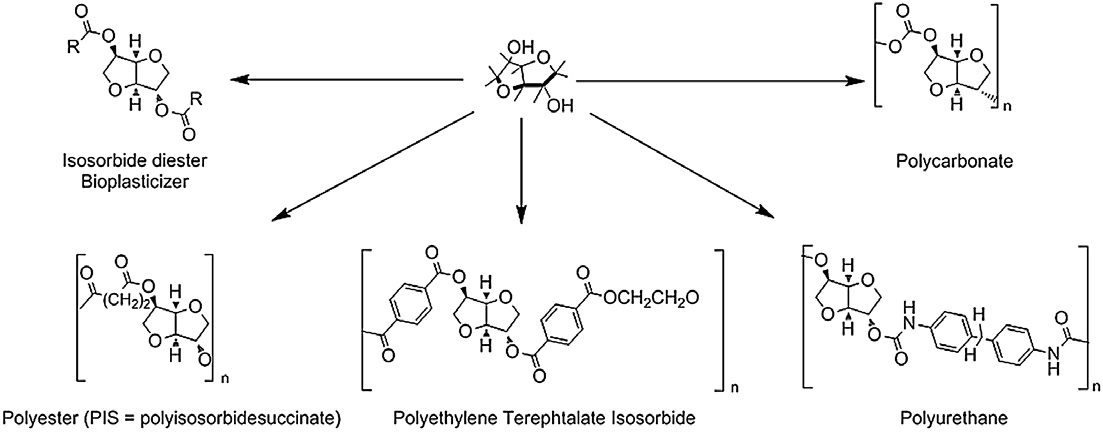
and are summarized in . As depicted, a lot of derivatives
1381-5148/$ - see front matter Ó 2013 Elsevier Ltd. All rights reserved.
V. Besse et al. / Reactive & Functional Polymers 73 (2013) 588–594
Scheme 1. Isosorbide based polymers.
could be synthesized such as polyesters polytriazoles
2. Experimental section
and polycarbonates A recent patent reports the syn-thesis of polycarbonates from isosorbide. However, few works deal
with PUs synthesis from isosorbide . A first study was reportedby Dirlikov and Schneider . Then other teams reported the use
Isosorbide, epichlorohydrin, lithium bromide and 1,10-diamin-
of isosorbide for the synthesis of PUs with high glass transition tem-
odecane were purchased from Sigma Aldrich and used as received.
perature (Tg) values, from 80 to 240 °C (
N,N-dimethylformamide (DMF) was purchased from SDS Carlo
) These works show that isosorbide leads to rigid
Erba (Val de Reuil, France). Before use, DMF was dried according
PUs with Tg values higher than those obtained from other diols such
to current methods, distilled and stored under argon atmosphere.
as butanediol, neopentyl glycol, dihydroxymethylcyclohexane) and
Deuterated solvents (CDCl3 and DMSO-d6) were purchased from
reveals the interest of isosorbide for industrial PUs.
Eurisotop (Saint-Aubin, France).
These results led Cognet-Georjon et al. to use isosorbide for rigid
segments of PUs with longer diols such as hydroxylated polybuta-
diene for the soft segments . Most of PUs from isosorbide aresynthesized by reaction between isosorbide and methylenediphe-
2.2.1. Epoxy index determination
nyl-4,40-diisocyanate (MDI) which is a carcinogenic mutagenic rep-
The epoxy index (EI) is defined by the mass of monomer con-
rotoxic (CMR) compound This drawback reduces the interest
taining one mole of epoxide group. This index was determined
of using isosorbide as a biobased diol and led to the synthesis of
by an acid–base titration. Epoxide groups were ring-opened by
PHUs thereof. Moreover, PU materials for coatings should have a
an excess of hydrochloric solution and this excess was determined
by titration with a sodium hydroxide solution with phenolphtha-
g around or below 0 °C which is not described in the literature.
The synthesis of PHUs from step growth polyaddition of dicy-
lein as a color indicator.
clocarbonates and diamines was extensively reported in literature,particularly by Endo Indeed, several cyclocarbonates
2.2.2. Isosorbide diglycidyl ether (E1)
were synthesized and some PHUs were thereof characterized
In a two necks round bottom flask equipped with an addition
Several methods are used to synthesize five-membered cyclic
funnel, isosorbide (8.5 g, 37.6 mmol) and chloroform (50 mL) were
carbonates . Most of these methods are based on epoxide
introduced. Then, m-chloroperbenzoic acid (47.2 g, 273.4 mmol)
or diol reactants. This is also the case of dicyclocarbonate syntheses
solubilized in chloroform (200 mL) beforehand, was introduced in
. Two bis and polycarbonates families were reported
the funnel and added dropwise. The mixture was stopped after
in the literature. The first one is constituted of carbonate esters
16 h when no more double bond signals were found in 1H NMR.
from glycerin carbonate, the second one is constituted of carbonate
The organic layer was washed three times: first with Na2SO3, then
ethers from glycidyl ether carbonatation In a recent paper,
Na2CO3 and water. Organic layers were put together and the sol-
Brocas et al. have reported the synthesis of cyclocarbonates
vent removed under reduced pressure. The crude was purified by
using mild conditions to synthesize crosslinked polyethers
column chromatography under silica (eluent:ethyl acetate). Evap-
Cyclocarbonate groups were
oration of the solvent with a rotary evaporator gave the corre-
synthesized by carbonatation of the corresponding epoxide groups
sponding product (5 g, 14.2 mmol) as yellowish viscous oil.
using a low carbon dioxide pressure (1 bar) and a catalytic amount
of lithium bromide (LiBr) at 80 °C.
1H NMR (400.1 MHz, CDCl3, d): 2.50–2.58 (m, 2H, H1a & H2a);
Our work aims to synthesize new cyclocarbonate from isosor-
2.72–2.76 (m, 2H, H1b & H2b); 3.04–3.17 (m, 2H, H2a & H11a);
bide by carbonatation. The originality of this work consists not
3.29–3.42 (m, 2H, H2b & H11b); 3.51–3.59 (m, 2H, H3a & H10a);
only in the synthesis of isosorbide dicyclocarbonate but also in
3.72–3.81 (m, 2H, H3b & H10b); 3.85–4.05 (m, 6H, Hisosorbide cycle);
the elaboration of original building block to synthesize low Tg
4.46 (ddt, 1H, 3JHH = 4.4 Hz, 3JHH = 11.6 Hz, 4JHH = 1.2 Hz, H6); 4.60
PHU, designed for coating applications, whereas isosorbide was
(ddt, 1H, 3JHH = 4.4 Hz, 3JHH = 8.4 Hz, 4JHH = 1.6 Hz, H7).
exclusively used for high T
g in literature. Moreover, we studied
C NMR (100.6 MHz, CDCl3, d): 44.4 (C1 & C12); 50.9 (C2 & C11);
the thermal degradation of PHUs and the compounds resulting of
70.4 (C5 & C8); 73.5 (C3 & C10); 80.6 (C4 & C9); 86.0 (C6 & C7).
degradation, which was never studied to the knowledge of theauthors. To the best of our knowledge, no paper or even patent re-
2.2.3. Isosorbide diglycidylether oligomers (E2 and E3)
ports the synthesis of these new PHUs, synthesized according the
In a round bottom flask equipped with an addition funnel and a
concept of ‘‘green chemistry''.
reverse Dean Stark surmounted by a condenser, isosorbide (1 eq.)
V. Besse et al. / Reactive & Functional Polymers 73 (2013) 588–594
and epichlorohydrin (10 eq.) were introduced. A sodium hydroxide
3. Results and discussion
solution (2.3 eq.) in water (50/50:wt/wt) was prepared and placedin the addition funnel. The mixture was heated at 115 °C under ar-
Firstly, isosorbide was converted into epoxidized oligoisosor-
gon atmosphere. The sodium hydroxide solution was added drop-
bide and then into cyclocarbonate oligoisosorbide by a carbonata-
wise. The reaction was stopped when no longer water was
tion reaction. Then the corresponding PHUs were synthesized by
collected in the Dean Stark apparatus, after approximately 6 h.
reaction with synthesized dicyclocarbonates and diamines. Their
The mixture was then allowed to stay at room temperature and
spectral and physical characterizations as well as the formation
epichlorohydrin was added to precipitate NaCl. After filtration,
of the polymer network were investigated. The last part of the arti-
the epichlorohydrin was removed by evaporation under reduced
cle is dedicated to the degradation study of PHUs by performing
pressure to give the desired product as yellowish viscous oil.
ATG-IR analyses in order to analyze the released products.
Yield: 98% (epoxide/isosorbide = 1.24). Dosage: 216 g/eq. epox-
ide rating: 4.63 meq/g (monomer).
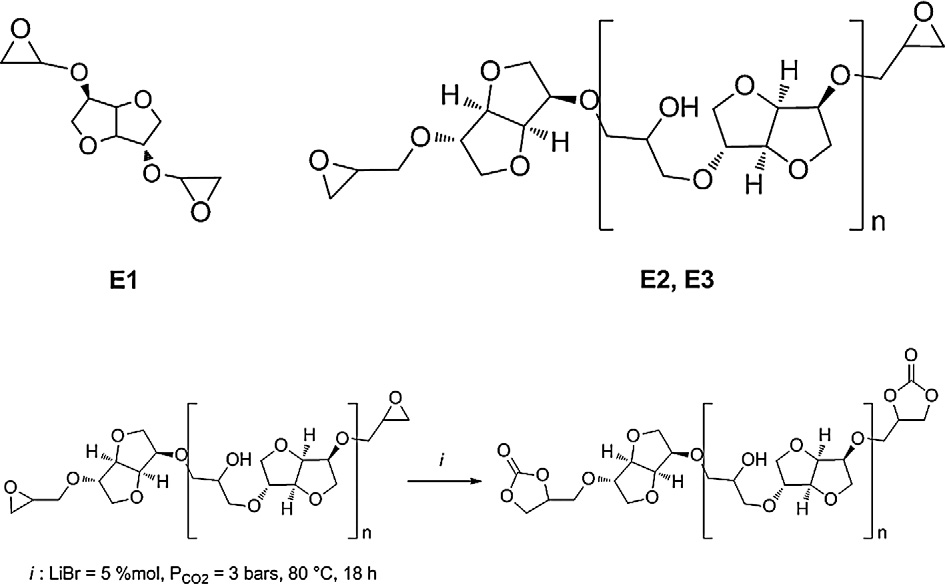
3.1. Synthesis of isosorbide diglycidylether oligomers (E1–3)
2.2.4. Carbonatation reaction: general procedure for DC1–DC3
This part focuses on the synthesis of diglycidylether from three
An epoxidized isosorbide (10.0 mmol of epoxide groups) and
different isosorbide reactants with various lengths. Epoxidized iso-
LiBr (0.5 mmol, 5% mol) were dissolved in dimethylformamide
sorbide E1 was synthesized by allylation of isosorbide followed by
(DMF, 45 mL). The solution was introduced into a reactor and the
its epoxydation using m-chloroperbenzoic acid. Isosorbide oligo-
atmosphere was replaced with CO
mers E2 and E3 were obtained by reaction of epichlorohydrin with
2 (P = 6 bars). The solution was
then allowed to stand at 80 °C with continuous stirring during
isosorbide at room temperature. Monomers E2 and E3 are different
12 h. DMF was removed by distillation (60 °C, P = 0.01 bar). The
by the length of the spacer between the two epoxide functions;
pure product was obtained quantitatively as yellowish highly vis-
this aspect will be discussed below ).
Two epoxidized oligoisosorbides E2 and E3 were obtained with
FTIR (m, cm�1) = 1780 (CO), 2860 and 2890 (CAC).
an epoxy index respectively of 4.13 meq/g and 2.9 meq/g (The ratio R of epoxy cycle to isosorbide cycle is given by the ratio
2.2.5. Polyhydroxyurethane (PHU1–PHU6) syntheses: general
between the integration of the epoxide protons (H1a–b and H2)
and the isosorbide protons (H�) following the method described
Step growth polyaddition of dicyclocarbonates (DC1–2) (1 eq.)
by Chrysanthos et al. Both ratio R obtained by NMR spectros-
was realized with diamines (1 eq.) and a catalyst (in a course of
copy and titration are in agreement either for epoxidized oligoiso-
patent registration) under stirring in distilled DMF (2 mL) and ar-
sorbide E2 and oligoisosorbide E3 (Chrysanthos et al.
gon atmosphere during 12 h at room temperature At the
mentioned other linear and branched oligomers. Branched oligo-
end of reaction, the formed polymers are still soluble. PHU1–6
mers can be explained by a competition during the reaction with
polymers, soluble in DMF, were precipitated in water (these PHUs
epichlorohydrin, between the secondary hydroxyl groups of isosor-
are not soluble in it). After drying by filtration then centrifugation,
bide and the secondary hydroxyl groups formed after a first reac-
PHU1–6 were quantitatively obtained. Both synthesized PHUs
tion of isosorbide diglycidyl ether with isosorbide. It means that
were characterized by 1H NMR spectrometry, FTIR analysis and
some oligomers can have more than two glycidyl groups.
SEC in DMF. Only 1H NMR spectra of the corresponding polyhydr-oxyurethane PHU2 is reported.
3.2. Synthesis of dicyclocarbonates DC1–3
FTIR (m, cm�1) = 1534 and 3082 (NH); 1694 (CO); 3328 (NH).
The dicyclocarbonates DC1–3 were synthesized by carbonata-
2.3. Analytical techniques
tion of isosorbide diglycidyl ether oligomers E1–3. This reactionwas carried out owing to Brocas et al. conditions during
All Nuclear magnetic resonance (1H NMR) measurements were
18 h. Total conversion was confirmed by the total disappearance
recorded on a Brucker AC-400 MHz spectrometer at room temper-
of the protons of the epoxide moiety between 2.56 and 2.82 ppm
ature in deuterated chloroform (CDCl
and also by the appearance of signals corresponding to the carbon-
or dimethylsulfoxyde
(DMSO). The chemical shifts were reported in parts per million rel-
ate protons between 4.20 and 4.88 ppm
ative to tetramethylsilane.
IR spectra were recorded with a Nicolet 210 FT-IR spectrometer.
3.3. Polyurethanes synthesis (PHU1-6)
Differential scanning calorimetry (DSC) analyses were per-
formed under inert atmosphere with a calorimeter DSC 200F3 from
Then, step growth polyadditions of DC1–2 with commercial dia-
Netzsch calibrated with indium standard. The polymer was
mines were carried out in presence of a catalyst (in a course of pat-
weighted in an aluminum pan and consecutively placed in the
ent registration) and yielded PHU1–6 PHUs at room temperature
measurement heating cell. An empty pan was used as reference.
The various diamine backbones are expected to provide
All the samples were heated under inert atmosphere from �120
a large range of mechanical properties to the PHUs. This part is fo-
to 100 °C at a heating rate of 20 °C/min. Three runs were recorded
cused on the development of polymers for coatings so that one of
and the glass transition temperature (Tg) values were measured
the absolutely desirable milestones is a Tg below 0 °C. According to
during the second run and confirmed by a third run. Tgs were cal-
the theory, a more flexible carbon chain should provide a lower Tg.
culated at the inflexion point of the heat capacity jump.
The reaction between a cyclocarbonate and a diamine leads to two
Thermogravimetric analyses (TGAs) were performed using a
regioisomers depending on the carbonate position attacked by the
TGA Q50 W/MFC apparatus of TA Instruments under air and nitro-
amine. Isomers such as primary/primary alcohol, primary/second-
gen flow (25 ml/min) from room temperature to 500 °C at a heat-
ary alcohol and secondary/secondary alcohol were obtained .
ing rate of 5 °C/min�1. The analysis consisted in registering the
We firstly report the results obtained with oligomers DC2 and
weight loss of the sample as a function of temperature.
DC3 when polymerized with DA10. The analyses show the high
Size exclusion chromatography (SEC) was performed on a Var-
reactivity of the reactants, the mechanical properties of the PHUs
ian ProStar Model 210 equipped with an RI refractive index detec-
but also the effect of the oligomer length on these properties. These
tor. Two PLgel 5 lm MIXED-C 600 mm were used at 70 °C with a
two PHUs (PHU2–3) were characterized by 1H NMR, FTIR and SEC
0.8 mL min�1 flow rate of DMF, calibrated using PMMA standards.
V. Besse et al. / Reactive & Functional Polymers 73 (2013) 588–594
Scheme 2. Idealized structure of glycidylisosorbide oligomers and synthesis of isosorbidedicyclocarbonates by carbonatation.
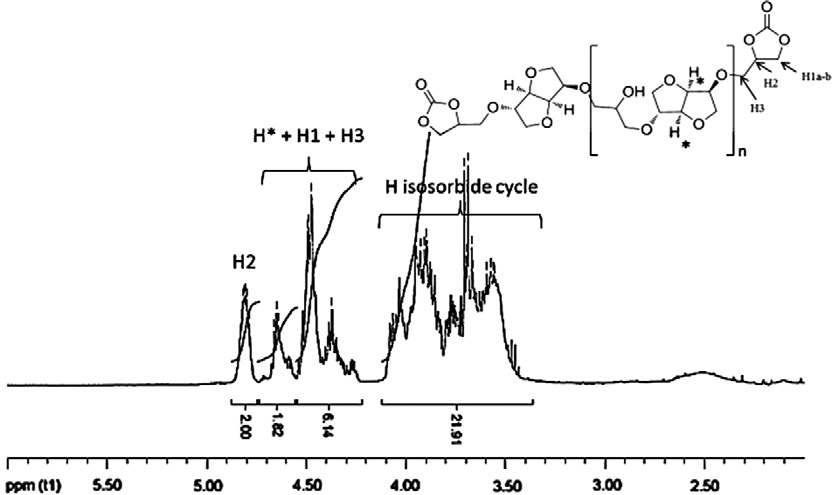
Fig. 1. 1H NMR spectrum of carbonated oligoisosorbide DC2.
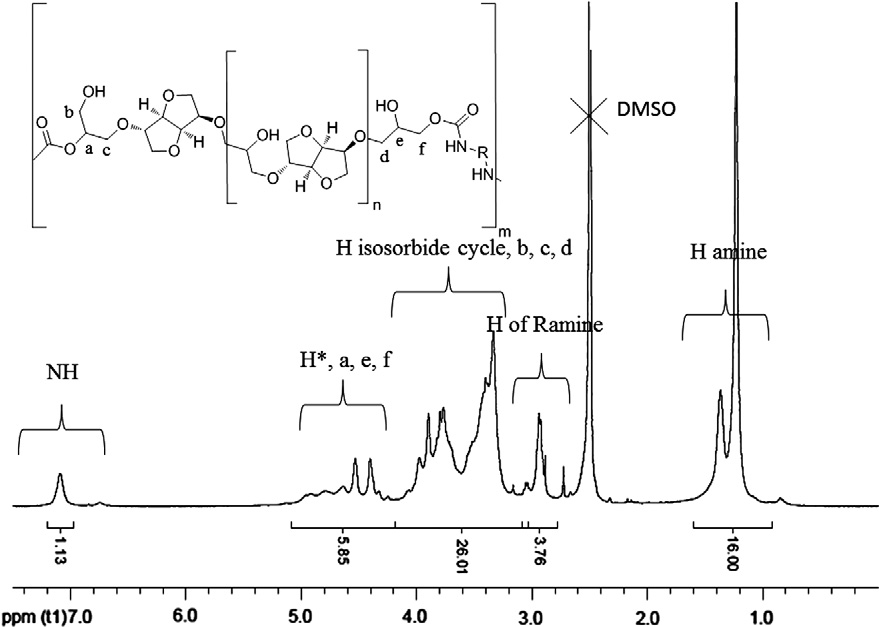
1H NMR spectrum of synthesized PHU2 is confirmed by the for-
mation of carbamate group with signal of proton HANCOO at
Determination of R using 1H NMR.
7.10 ppm and alcohol at 4.40 ppm
FTIR spectrum confirms the formation of PHU2 with the three
I (Integration value)
I (Integration value)
characteristic bands of carbamate function: NAH bond stretching
vibration, hydrogen bonded C@O stretching and NAH bond defor-
mation are respectively observed at 3082 cm�1, 1694 cm�1 and
H� (isosorbide cycle)
1534 cm�1. The large absorption of the hydroxyl group OH at
Ratio R (titration)
3328 cm�1 also appeared. Moreover, the absence of the absorption
band of the carbonyl of the carbonate group at 1785 cm�1 reveals a
Molar mass (g mol�1)
total conversion of initial carbonated isosorbide ().
Size exclusion chromatography (SEC) analyses have been per-
formed on PHU2 and PHU3. Molecular weights were found to be
Table of composition of PHU1-6.
quite low, Mn (7800 and 8600 g/mol for PHU2 and PHU3 respec-
tively) and PDI were quite high (2.6 and 6.3 for PHU2 and PHU3respectively) ).
The properties of these synthesized polyurethanes are similar to
those found for isosorbide based polyurethanes described by Cog-
net-Georjon et al. especially concerning the low value obtained
for Mn. The high value of PDI and low molecular weight is certainly
due the presence of initial branched oligomers which are insoluble
V. Besse et al. / Reactive & Functional Polymers 73 (2013) 588–594
Fig. 2. 1H NMR spectrum (400 MHz, DMSO-d6) of PHU2.
Table 3Thermal properties of PHU2 and PHU4-6.
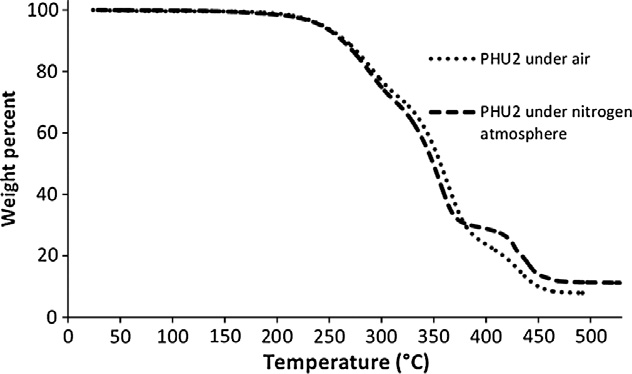
TGA analysis shows a 5% mass loss (Td 5%) at 243 and 237 °C for
PHU2 and PHU3 respectively. The thermal degradation with for-mation of volatiles of the two PHUs is similar and proceeds in threesteps under air as well as under nitrogen atmosphere
For example, for PHU2, the first step between 180 and 310 °C
Fig. 3. TGA spectrum of PHU2 under air and nitrogen atmosphere.
corresponds to a 28% weight loss. Then the second step, between342 and 379 °C corresponds to a 48% weight loss. The third step be-tween 420 and 451 °C corresponds to a 48% weight loss and theresidue represents 11 wt.% of the initial weight at 500 °C. In bothcases, no difference between oxidative or inert atmosphere wasfound so that no oxidation phenomenon occurs. The weakest bondis supposed to be the urethane one (EC–NH = 98 kJ/mol) and its deg-radation is supposed to be the same under oxygen and nitrogenatmosphere
The next part of this article is devoted to the study of the dia-
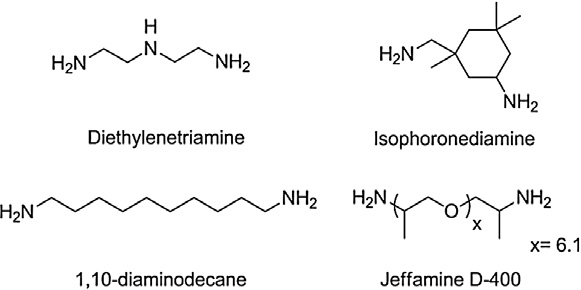
mine structure effect. Since the oligomer length shows no evidencein modifying the final polymer properties, the next part of thestudy will focus on PHUs obtained from DC2 and three other dia-mines e.g. Jeffamine D-400, diethylenetriamine and isophoronedi-amine and
Scheme 3. Structures of amines used in this study.
We first studied the thermal properties (Td 5% and Tg) of PHU4–
6. Due to the rigid structure of IPDA, the Tg obtained for PHU4
in DMF and can interact intensively with the GPC column. Both
(59 °C) is higher than the one obtained with DA10 and other dia-
PHU2 and PHU3 are amorphous as determined by DSC analysis
mines. Then DETA, a soft aliphatic triamine, allows PHU5 to reach
(They exhibit similar T
g of 0 °C. The last amine, Jeffamine D-400, is a long polyether
15 and 19 °C respectively. Compared to the previous results found
chain diamine which confers high flexibility and toughness to
in the literature (), and especially
PHU6 so that its Tg is the lowest (�8 °C)
those with the aliphatic diisocyanate, hexamethylene diisocyanate
This result is compatible with a use in coatings. DMF and DMSO
(HDI), the first addition of glycidyl functions on the isosorbide be-
appear to be the best solvent as all materials are soluble in it
fore the synthesis of bis-carbonate can explain the lower T
whereas none of them are soluble in water (
served and the increase softness of PHUs.
V. Besse et al. / Reactive & Functional Polymers 73 (2013) 588–594
Fig. 4. ATG-IR analysis of PHU2 and PHU4.
The last part of our study is dedicated to the characterization of
Linear and branched PHUs were obtained with Tg values in the
the released compounds during the degradation process of PHUs
range from �8 to 59 °C, and low Tg PHUs are suitable for coatings
by the ATR-IR technique The decomposition of urethane
application. An acceptable thermal stability (Td between 234 and
bonds is depending on the structures of the reactants which were
255 °C) was determined owing to TGA for all PHUs. This work dem-
used for their synthesis. If the first decomposition is due to hard
onstrates that biobased PHUs can be obtained easily from isosor-
segments, the second and third decompositions were attributed
bide as hard segment and with ether bonds. Works are in
to soft segments. Javni et al. have demonstrated that the first deg-
progress for a better control of molecular weight and polydisper-
radation step for polyurethanes from functionalized vegetable oils
sity. Last but not least, the compounds released by the degradation
was the production of carbon dioxide. This carbon dioxide was sus-
of PHUs were analyzed and found to be carbon dioxide and second-
pected to come from the formation of carbodiimides but more
ary amines. Interestingly, these new PHUs do not release isocya-
credibly from the disruption of the urethane bond following mech-
nates during thermal degradation. Choices of other aliphatic
anisms 2 and 3 .
biobased diamines, shorter one like C5 or longer one like C36 will
PHU2 and PHU4 were typically introduced into an ATG instru-
permit to prepare hard and soft PHUs.
ment and the released gases were analyzed by an infra-red appara-tus after a given time ). After only 495s, the first degraded
bond is the carbonyl of the urethane function (mCOurethane = 1720 -cm�1) as expected Carbon dioxide and secondary amine bands
This work was supported by ANR project GreenCoat. The
(mCO = 670, 2320, 3700 cm�1 and m
NH = 1095, 1490, 3800 cm�1).
authors also thank Specific Polymers for the synthesis of isosorbide
Alkane groups, probably PHUs spacers, were detected by the
diglycidyl ether compounds under the reference SP 9S-5-001.
presence of two large bands between 2800 and 3000 cm�1. Exceptthe carbonyl urethane group, the same bands can be observedalong the thermal analysis (i.e. CO2 and NH bands). PHU4 gave
Appendix A. Supplementary material
same results as PHU2 concerning the degradation products. More-over, we can clearly see that no isocyanate, alcohol or alkene were
Supplementary data associated with this article can be found, in
formed during the degradation process so that we can clearly dem-
the online version, at
onstrate that PHUs degradation products are CO2 and secondary
amines. This result is of importance since it shows that not onlyPHUs are synthesized without isocyanates, but also they do not
lead to isocyanate during thermal degradation.
[1] M.H. Karol, J.A. Kramarik, Toxicol. Lett. 89 (1996) 139–146.
[2] H. Tomita, F. Sanda, T. Endo, J. Polym. Sci. Part A. Polym. Chem. 39 (2001) 860–
Isosorbide was functionalized with glycidyl ether groups and
[3] H. Tomita, F. Sanda, T. Endo, J. Polym. Sci. Part A. Polym. Chem. 39 (2001)
the quantity of epoxide groups was determined by a titration. Re-
[4] H. Tomita, F. Sanda, T. Endo, J. Polym. Sci. Part A. Polym. Chem. 39 (2001) 162–
sults obtained by titration were in agreement with those obtained
by 1H NMR spectroscopy. Then functionalized oligoisosorbides
[5] W. Ried, W. Merkel, Angew. Chem. Int. Ed. English 8 (1969) 379–380.
[6] H. Tomita, F. Sanda, T. Endo, J. Polym. Sci. Part A. Polym. Chem. 39 (2001)
were carbonated for the first time using mild conditions with a to-
tal conversion. This work led to the synthesis of new biobased
[7] C.D. Diakoumakos, D.L. Kotzev, WO Patent 2005016993, 2005.
PHUs using the cyclocarbonate-aliphatic amine chemistry in the
[8] O. Figovsky, L. Shapovalov, Macromol. Symp. 187 (2002) 325–332.
[9] O. Figovsky, L. Shapovalov, N. Blank, F. Buslov, US Patent 2004192803, 2004.
presence of a catalyst. This reaction was found to be very effective
[10] S. Benyahya, M. Desroches, R. Auvergne, S. Carlotti, S. Caillol, B. Boutevin,
as the reaction of cyclocarbonate groups is completed within 12 h.
Polym. Chem. 2 (2011) 2661–2667.
V. Besse et al. / Reactive & Functional Polymers 73 (2013) 588–594
[11] S. Benyahya, J.-P. Habas, R. Auvergne, V. Lapinte, S. Caillol, Polym. Int. (2012)
[31] R. Marín, A. Alla, A. Martínez de Ilarduya, S. Muñoz-Guerra, J. Appl. Pol. Sci. 123
(2012) 986–994.
[12] M. Desroches, S. Caillol, R. Auvergne, B. Boutevin, G. David, Polym. Chem. 3
[32] H.K. Lindberg, A. Korpi, T. Santonen, K. Säkkinen, M. Järvela, J. Tornaeus, N.
(2012) 450–457.
Ahonen, H. Järventaus, A.L. Pasanen, C. Rosenberg, H. Norppa, Mut. Res. J. 1
[13] F. Fenouillot, A. Rousseau, G. Colomines, R. Saint-Loup, J.P. Pascault, Prog.
(2011) 1–10.
Polym. Sci. 35 (2010) 578.
[33] D. Tang, D.J. Mulder, B.A.J. Noordover, C.E. Koning, Macromol. Rapid Commun.
[14] M. Rose, R. Palkovits, Chem. Sus. Chem. 5 (2012) 167.
32 (2011) 1379–1385.
[15] E. Cognet-Georjon, F. Mechin, J.P. Pascault, Macromol. Chem. Phys. 196 (1995)
[34] A. Steblyanko, Polym. Chem. 38 (2000) 2375–2380.
[35] N. Kihara, T. Endo, J. Polym. Sci. Part A. Polym. Chem. 31 (1993) 2765–2773.
[16] P. Tendo, F. Arico, G. Gauthier, L. Rossi, A.E. Rosamilia, H.S. Bevinakatti, R.L.
[36] M.R. Kim, H.S. Kim, C.S. Ha, D.W. Park, J.K. Lee, J. Appl. Polym. Sci. 81 (2001)
Sievert, C.P. Newman, Chem. Sus. Chem. 3 (2010) 566–570.
[17] S. Thiyagarajan, L. Gootjes, W. Vogelzang, J. Van Haveren, M. Lutz, D.S. Van Es,
[37] L. Ubaghs, Macromol. Rapid Commun. 25 (2004) 517–521.
Chem. Sus. Chem. 4 (2011) 1823–1829.
[38] Q. Li, W. Zhang, N. Zhao, W. Wei, Y. Sun Catalysis. Today. 115 (2006) 111–116.
[18] S. Thiyagarajan, L. Gootjes, W. Vogelzang, Tetrahedron 67 (2011) 383–389.
[39] Z. Fang, CN101376632A, 2009.
[19] J. Pfeffer, M. Ortlet, E. Spyrou, T. Hass, U. Korek, H. Schmidt, U. Dingerdissen,
[40] T. Nishikubo, T. Iizawa, M. Iida, Tetrahedron Lett. 27 (1986) 3741–3744.
WO 2011/000585 A1, 2011.
[41] M. Aresta, A. Dibenedetto, C. Dileo, I. Tommasi, E. Amodio, J. Supercrit. Fluids
[20] J.C. Bersot, N. Jacquel, R. Saint-Loup, P. Fuertes, A. Rousseau, J.P. Pascault, R.
25 (2003) 177–182.
Spitz, F. Fenouillot, V. Monteil, Macromol. Chem. Phys. 212 (2011) 2114–
[42] B.M. Bhanage, S.I. Fujita, Y. Ikushima, Green Chem. 5 (2003) 429–432.
[43] H. Komura, T. Yoshino, Y. Ishido, Bull. Chem. Soc. Jap. 46 (1973) 550–553.
[21] C. Besset, S. Binault, M. Ibert, P. Fuertes, J.P. Pascault, E. Fleury, J. Bernard, E.
[44] K. Weissermel, Industrial Organic Chemistry, third ed., Wiley-VCH, 1997.
Drockenmuller, Macromolecules 43 (2010) 17–19.
[45] G. Proempers, H. Keul, H. Hoecker Des, Monomers Polym. 8 (2005) 547–569.
[46] S.P. Rannard, N.J. Davis, Org. Lett. 1 (1999) 933–936.
Biomacromolecules 11 (2010) 2797–2803.
[47] K.T. Sprott, E.J. Corey, Org. Lett. 5 (2003) 2465–2467.
[23] C. Besset, J. Bernard, E. Fleury, J.P. Pascault, P. Cassagnau, E. Drockenmuller,
[48] A.L. Brocas, G. Cendejas Santana, S. Caillol, A. Deffieux, S. Carlotti, J. Polym. Sci.
R.J.J. Williams, Macromolecules 43 (2010) 5672–5678.
Part A. Polym. Chem. 49 (2011) 2677–2684.
[24] M. Fuji, M. Akita, T. Tanaka, Mitsubishi Chem.: EP 2 033 981 A1, (2009).
[49] A. Soules, S. Caillol, B. Boutevin, J.-P. Joubert, J. Martins. Patent registration
[25] B.J.P. Jansen, J.H. Kamps, H. Looij, E. Kung, WO 2009/052463 A1, 2009.
number 1159818, 28/10/2011.
[26] M. Kinoshita, M. Saito, K. Hironaka, EP 2149 589 A1, (2010).
[50] M. Chrysanthos, J. Galy, J.P. Pascault, Polymer 52 (2011) 3611–3620.
[27] P. Fuertes, M. Ibert, E. Josien, P. Tundo, F. Arico, WO 2011/039483 A1, 2011.
[51] A. Steblyanko, W. Choi, F. Sanda, T. Endo, J. Polym. Sci. Part A: Polym. Chem. 38
[28] S.K. Dirlikov, C.J. Schneider, US4443563, 1984.
(2000) 2375–2380.
[29] C.H. Lee, H. Takagi, H. Okamoto, M. Kato, A. Usuki, J. Polym. Sci. Part A. Polym.
[52] B. Boutevin, J.P. Hugon, Y. Pietrasanta Eur, Polym. J. 17 (1981) 723–727.
Chem. 47 (2009) 6025–6031.
[53] K. Ashida (Ed.), Taylor & Francis group, CRC Press, Boca Raton, USA, 2007, pp.
[30] E. Cognet-Georjon, F. Mechin, J.P. Pascault, Macromol. Chem. Phys. 197 (1996)
[54] I. Javni, Z.S. Petrovic, A. Guo, R. Fuller, J. Appl. Polym. Sci. 77 (2000) 1723–1734.
Source: http://virtualpu.com/uploads/user_product_attachments/0-2015-12-09%2012:38:36-2014-2.pdf
DOCUMENTOS DE LICITACIÓN No. 1506 – 210011 ALCANCE DE LA LICITACIÓN El Comprador es: Fondo Financiero de Proyectos de Desarrollo – FONADE. El nombre y número de identificación de la LPN son: "Adquisición de tres (3) equipos de laboratorio (GPHF-Minilab) con stock de patrones de análisis para apoyar la evaluación de calidad de los antimaláricos en los laboratorios de salud pública de los Departamentos de Chocó, Córdoba y Cauca", Licitación Pública Nacional No. 1506 – 210011. 2.
Guía de Práctica Clínica sobre el Manejo de la Depresión Mayor en el Adulto GUÍAS DE PRÁCTICA CLÍNICA EN EL SNSMINISTERIO DE SANIDAD Y CONSUMO Guía de Práctica Clínica sobre el Manejo de la Depresión Mayor en el Adulto GUÍAS DE PRÁCTICA CLÍNICA EN EL SNSMINISTERIO DE SANIDAD Y CONSUMO Esta GPC es una ayuda a la toma de decisiones en la atención sanitaria. No es de obligado cumplimiento ni sustituye al juicio clínico del personal sanitario.