Zen99974.zen.co.uk
Long chain polyunsaturated fatty acids are required
for efficient neurotransmission in C. elegans
Giovanni M. Lesa1,*,‡, Mark Palfreyman2, David H. Hall3, M. Thomas Clandinin4, Claudia Rudolph5,
Erik M. Jorgensen2 and Giampietro Schiavo1
1Molecular Neuropathobiology Laboratory, Cancer Research UK, London Research Institute, Lincoln's Inn Fields Laboratories, 44 Lincoln's Inn
Fields, London WC2A 3PX, UK
2Department of Biology, University of Utah, Salt Lake City, Utah 84112-0840, USA
3Center for C. elegans Anatomy, Department of Neuroscience, Albert Einstein College of Medicine, Bronx, New York 10461, USA
4Nutrition and Metabolism Research Group, University of Alberta, Edmonton, Alberta, T6G 2P5, Canada
5EleGene AG, Am Klopferspitz 19, 82152 Martinsried, Germany
*Present address: MRC Laboratory for Molecular Cell Biology, University College London, Gower Street, London WC1E 6BT, UK
‡Author for correspondence: (e-mail:
[email protected])
Accepted 2 October 2003Journal of Cell Science 116, 4965-4975 2003 The Company of Biologists Ltddoi:10.1242/jcs.00918
The complex lipid constituents of the eukaryotic plasma
Expression of functional fat-3 in neurons, or application
membrane are precisely controlled in a cell-type-specific
exogenous LC-PUFAs to adult animals rescues
manner, suggesting an important, but as yet, unknown
defects. Pharmacological, ultrastructural and
cellular function. Neuronal membranes are enriched in
electrophysiological analyses demonstrate that fat-3
long-chain polyunsaturated fatty acids (LC-PUFAs) and
mutant animals are depleted of synaptic vesicles and
alterations in LC-PUFA metabolism cause debilitating
release abnormally low levels of neurotransmitter at
neuronal pathologies. However, the physiological role of
cholinergic and serotonergic neuromuscular junctions.
LC-PUFAs in neurons is unknown. We have characterized
These data indicate that LC-PUFAs are essential for
the neuronal phenotype of C. elegans mutants depleted of
efficient neurotransmission in C. elegans and may account
LC-PUFAs.
for the clinical conditions associated with mis-regulation of
The C. elegans genome encodes a single ∆
6-desaturase
LC-PUFAs in humans.
gene (fat-3), an essential enzyme for LC-PUFA
biosynthesis. Animals lacking fat-3 function do not
synthesize LC-PUFAs and show movement and egg-laying
Key words:
C. elegans, Neuromuscular junction, Neurotransmitter
abnormalities associated with neuronal impairment.
release, Polyunsaturated fatty acids, Synapse
functions of these molecules we generated
Caenorhabditis
One of the central challenges in biology is to understand the
elegans mutants depleted of LC-PUFAs and analyzed their
cellular functions of the wide variety of complex lipids present
in animal cells. Long-chain polyunsaturated fatty acids (LC-
The nematode
C. elegans synthesizes many of the LC-
PUFAs), fatty acids with multiple double bonds, are
PUFAs found in humans (Wallis et al., 2002) and represents a
synthesized from dietary precursors and are localized to cell
good model organism to systematically study the neuronal
membranes as phospholipid esters. Both the absolute LC-
roles of these molecules. First, the location, structure and
PUFA levels and their relative concentrations are strictly
function of virtually all
C. elegans neurons are known. Second,
controlled in mammalian neurons (Lauritzen et al., 2001)
pharmacological assays can be used to test synaptic function
implying that LC-PUFAs have critical neuronal functions.
of certain neurons such as cholinergic motor neurons and
Indeed, mutations in enzymes involved in LC-PUFA
serotonergic neurons (see Jorgensen et al., 1995; Lackner et al.,
metabolism cause a form of X-linked mental retardation
1999; Miller et al., 1999). Third, it is possible to study the role
(Meloni et al., 2002) and two forms of macular dystrophy
of LC-PUFAs independently of the known eicosanoid-
(Zhang et al., 2001) and diets deficient in essential LC-PUFAs
mediated signaling pathways, since the
C. elegans genome
are associated with deficits in infant brain function (Anderson
does not apparently encode orthologs of cyclooxygenase,
et al., 1999; Helland et al., 2003; Lauritzen et al., 2001; Willatts
lipooxygenase, thromboxane synthase or any orthologs of
et al., 1998). In addition, LC-PUFAs induce oligomerization of
prostaglandin, leukotriene or thromboxane receptors. Finally,
α-synuclein, a protein found as insoluble aggregates in α-
C. elegans mutants depleted of LC-PUFAs have been isolated
synucleinopathies including Parkinson's disease (Sharon et al.,
by inactivating the gene
fat-3, which encodes ∆6-desaturase,
2003). Although these and other studies suggest an important
an enzyme essential for LC-PUFA biosynthesis (Fig. 1; this
role of LC-PUFAs in nervous system function, their precise
study and Watts and Browse (Watts and Browse, 2002)). Initial
role in neurons remains unclear. To address the neuronal
analysis revealed that some of the defects displayed by
fat-3
Journal of Cell Science 116 (24)
mutants are suggestive of neuronal impairment (Watts et al.,
Materials and Methods
2003), yet the molecular basis of these effects are unknown.
Strains and isolation of fat-3 deletions
Here we characterize in detail the neuronal phenotypes
C. elegans strains were cultured at 20°C as described (Brenner, 1974).
displayed by
fat-3 mutants and demonstrate that LC-PUFAs are
The strain GR1333 (
IsPtph-1::gfp) was provided by I. Y. Sze and G.
essential for normal neurotransmitter release at cholinergic and
Ruvkun (University of California Irvine, CA and Harvard University,
serotonergic neuromuscular junctions (NMJs). These defects
Cambridge, MA) (Sze et al., 2000).
are not developmental but are functional, since exogenous LC-
To isolate deletion of the
fat-3 gene, we constructed DNA libraries
PUFAs can rescue mutant adults. Consistent with these deficits
of approximately 5,400,000 haploid genomes from wild-type (N2)
we provide pharmacological and electrophysiological evidence
animals mutagenized with ethylmethane sulfonate or with trimethylpsoralen. Using the polymerase chain reaction (PCR) with
fat-3
that animals lacking LC-PUFAs release abnormally low levels
primers (Jansen et al., 1997) we isolated two deletions:
fat-3(lg8101)
of neurotransmitter. In addition, ultrastructural analysis reveals
and
fat-3(qa1811). Both deletions confer a fully recessive phenotype.
that synapses in these animals are severely depleted of synaptic
We used PCR-based genotyping to verify that the deleted strains do
vesicles. We conclude that LC-PUFAs are required for efficient
not contain duplications of the intact
fat-3 gene. Analyses were carried
out in
fat-3(lg8101) homozygous, in
fat-3(lg8101)dpy-20(e1282)/unc-24(e138)fat-3(qa1811)dpy-20(e1282) heterozygous or in
fat-3(wa22)homozygous animals.
fat-3 mutants were outcrossed to wild-type
animals at least six times before analysis.
Total RNA for RT-PCR was extracted from mixed
C. elegans
populations (Chomczynski and Sacchi, 1987). 1 µg of total RNA was
reverse transcribed and PCR amplified using Ready-to-go RT-PCR
beads (Amersham Biosciences, Chalfont St. Giles, UK). RT-PCR for
unc-22 mRNA was used as positive control (data not shown). Primers
used (Fig. 2A): a, 5′-CTCGAATTTTAAACAACTTCGCCGC-3′; b,5′-GGCAGCTTTAGCTTGAATGTGCTC-3′; c, 5′-CAGAAGCTTC-
Rescue experiments
A region comprising 1 kb 5′ of the
fat-3 start, the entire
fat-3 codingsequence (Napier et al., 1998) and 0.9 kb 3′ of the
fat-3 end rescuedthe defects associated with
fat-3 mutants. Primers containing
KpnI and
SacI restriction sites were used to PCR amplify the entire
fat-3 coding
sequence. This
KpnI-
SacI fragment and a
HindIII-
KpnI fragment from
Punc-119::gfp (Maduro and Pilgrim, 1995) were cloned intopPD49.26 (provided by A. Fire, Carnegie Institution of Washington,
Baltimore, MD) to generate
Punc-119::fat-3. To generate
Pmyo-
3::fat-3, a
HindIII-
BamHI fragment including the
myo-3(+) promoter
and the
KpnI-
SacI PCR fragment including the entire
fat-3 coding
sequence were cloned into pPD49.26. This
KpnI-
SacI
fat-3 fragment
and a
PstI-
SmaI fragment containing the
elt-2 promoter were cloned
into pPD49.26 to generate
Pelt-2::fat-3. Constructs were injected
(Mello et al., 1991) at 1-100 ng/µl in
dpy-20(e1282) backgrounds with
dpy-20(+) pMH86 (20 ng/µl). The presence of
fat-3 mRNA was
verified in all transgenic lines by RT-PCR (data not shown).
Dp TGCTTGCAT.
Fig. 1. fat-3 encodes a ∆6-desaturase. (A) The LC-PUFA synthetic
pathways (Lauritzen et al., 2001; Spychalla et al., 1997). ALA, α-
linolenic acid; LIN, linoleic acid; DGLA, dihomo-γ-linolenic acid;
EPA, eicosapentaenoic acid; AA, arachidonic acid; DHA,docosahexaenoic acid. (B) The
fat-3 gene (W08D2.4) is located on
chromosome IV, between
unc-24 and
dpy-20. A 4.7 kb genomic
fragment including 977 bp 5′ and 862 bp 3′ of the
fat-3 coding region
rescues
fat-3 mutants. The coding regions are in boxes and the non-
coding regions are shown as lines. The cytochrome b5-like domain is
in gray. Asterisks indicate histidine-rich regions. The
fat-3(lg8101)
and
fat-3(qa1811) deletions and their breakpoints are shown. T
indicates an A to T substitution. Dp indicates a 17 bp duplication
(GAAAATGGTTGAATCAT).
fat-3(wa22) is a C to T point mutationthat changes S186 to F. (C) ClustalX alignment of FAT-3 protein with
human and plant (
Borago officinalis) ∆6-desaturases. The triangle
indicates the
fat-3(wa22) mutation
. Single-letter abbreviations for
amino acid residues are used. Identical and similar amino acids are
identified by gray and light gray shading, respectively. The putative
cytochrome b5-like domain is indicated with a line. Histidines
important for catalytic activity are marked by asterisks.
LC-PUFAs and neurotransmission
For fatty acid rescue experiments, arachidonic acid (AA),
between assays, wild-type controls were included in each assay. The
docosahexaenoic acid (DHA) or linoleic acid (LIN; Sigma, Poole,
acetylcholine (ACh) sensitivity was tested by spreading 1 M ACh on
UK) were prepared as 100 mg/ml solution in 95% ethanol and 100 µl
plates (final concentration 10 mM). To minimize ACh hydrolysis,
were spread over nematode growth medium plates.
E. coli OP-50 were
the assay was started within 10 minutes. 35-40 animals per experiment
then added as a food source and 3-20 L4-adult
fat-3(lg8101)dpy-
were scored for complete paralysis (Lackner et al., 1999) and 3-6
independent experiments per dose and per drug were carried out.
were grown on each plate. These hermaphrodites or their progeny
Statistical analysis was performed using the Mann-Whitney test with
(thus exposed to LC-PUFAs from hatching), were analyzed.
InStat software (GraphPad Software, San Diego, CA).
Motility, egg-laying and paralysis assays
Visualization of neurons and synapses
These assays were carried out in
fat-3(lg8101)dpy-20(e1282)/unc-
To visualize serotonergic neurons in
fat-3 mutants, we constructed
the strain
fat-3(lg8101);Is(Ptph-1::gfp,rol-6D).
tph-1 encodes a
fat-3(lg8101) mutants transgenic for the indicated constructs, or N2
tryptophan hydroxylase and is expressed in serotonergic neurons (Sze
animals. Motility was quantified by placing adult worms in the centre
et al., 2000). Animals were immobilized with 10 mM levamisole,
of a bacterial lawn on a Petri dish and allowing them to move. After
mounted on a 2% agarose pad and observed under a 40× or a 63×
30 seconds worms were immobilized with heat. Pictures of the tracks
objective on a Zeiss LSM510 confocal microscope (Carl Zeiss,
left on the bacterial lawn were taken using a Leica MZ 125
Oberkoken, Germany).
microscope (Leica Microsystems, Milton Keynes, UK) equipped with
For electron microscopy, young adult nematodes were fixed by
a Photometrics CoolSnap digital camera (Roper Scientific, Tucson,
immersion in buffered aldehydes and stained in osmium tetroxide
AZ) and Openlab software (Improvision, Coventry, UK). Single
(Hall, 1995) or fast frozen under high pressure followed by freeze
tracks were highlighted with Adobe Illustrator (Adobe Systems,
substitution into osmium tetroxide in acetone (McDonald, 1999).
Uxbridge, UK) and their pixels counted using NIH Image (National
Samples were embedded into plastic resin, thin sectioned using a
Institutes of Health, Bethesda, MD). 20-47 tracks from at least two
diamond knife, counterstained with uranyl acetate and lead citrate,
independent transgenic stable lines per genotype were analyzed.
and examined on a Philips CM10 electron microscope (Philips
Egg-laying-defective animals were determined by cloning L4
Electron Optics, Eindhoven, The Netherlands). We used 2 fixation
hermaphrodites to single plates and by scoring them every day for 4
conditions because
fat-3 mutants are, for some unknown reason,
days for embryos that hatched inside the mother.
difficult to fix well and the quality of the images obtained was not
The egg-laying assay in the presence of drugs was performed in
optimal. However, we found similar results with either fixation
microtiter wells as described previously (Trent et al., 1983) using
method. Both,
fat-3(lg8101) and
dpy-20(e1282)fat-3(lg8101)/unc-
serotonin or fluoxetine (Sigma) dissolved at the indicated doses in M9
24(e138)fat-3(qa1811)dpy-20(e1282) mutants displayed similarly
buffer. 12-36 animals for each dose were analyzed.
depleted synapses. Morphometric analysis was carried out in animals
For the paralysis assay, plates containing 1 mM aldicarb
stained with osmium tetroxide. Vesicles of approximately 30 nm
(Greyhound Chromatography, Birkenhead, UK) or 0.2 mM
diameter were counted. Statistical analysis was performed using
levamisole (Sigma) were prepared fresh for each set of assays as
SAS/STAT software (SAS Institute Inc., Cary, NC). Significance
described previously (Miller et al., 1999). To allow comparisons
values were calculated using Student's
t-test.
ACh and fatty acid quantificationACh was quantified in
fat-3(lg8101)dpy-20(e1282)/unc-24(e138)fat-3(qa1811)dpy-20(e1282) or
dpy-20(e1282) animals as describedpreviously (Nonet et al., 1993).
The genotypes of animals used for fatty acid quantification were as
follows:
fat-3(lg8101), N2, or
fat-3(lg8101)dpy-20(e1282);Ex(fat-3(+)dpy-20(+)). Lipids were extracted and partitioned (Hargreavesand Clandinin, 1988). Phospholipid-derived fatty acid methyl esterswere separated by capillary gas liquid chromatography using a fullyautomated Varian 6000 GLC (Varian Instruments, Mississauga,Ontario). Data were expressed as a percentage of the area count foreach individual fatty acid relative to all fatty acids combined.
ElectrophysiologyElectrophysiological methods were performed as previouslydescribed (Richmond et al., 1999; Richmond and Jorgensen, 1999)with minor adjustments. Briefly, the animals were immobilized in
Fig. 2. fat-3 deleted mutants do not synthesize
fat-3 mRNA.
cyanoacrylic glue and a lateral incision was made to expose the ventral
(A) Schematic of the
fat-3(lg8101) and
fat-3(qa1811) deletions and
medial body wall muscles. The preparation was then treated with
the primers used for
fat-3 mRNA analysis. (B,C) No wild-type
fat-3
collagenase (type IV; Sigma) for 15 seconds at a concentration of 0.5
mRNA is detected in
fat-3(lg8101) or
fat-3(lg8101)/fat-3(qa1811)
mg/ml. The muscle was then voltage clamped using the whole cell
mutant animals. (B) RT-PCR of total mRNA from
fat-3(lg8101) or
configuration at a holding potential of –60 mV. All recording were
wild-type animals with primers a and b, in the presence (+) or in the
made at room temperature (21°C) using an EPC-9 patch-clamp
absence (–) of reverse transcriptase. Predicted PCR products: cDNA,
amplifier (HEKA, Southboro, MA) run on an ITC-16 interface
281 bp; genomic DNA, 326 bp. (C) RT-PCR with primers b and c of
(Instrutech, Port Washington, NY). Data were acquired using Pulse
total mRNA from
fat-3(lg8101), wild-type or
fat-3(lg8101)/fat-
software (HEKA).
3(qa1811) animals. Predicted PCR products: cDNA, 999 bp;
The extracellular solution contained: 150 mM NaCl, 5 mM KCl,
genomic DNA, 1,451 bp.
0.5 mM CaCl2, 4 mM MgCl2, 10 mM glucose, 15 mM Hepes, pH
Journal of Cell Science 116 (24)
7.35, and sucrose to 340 mOsm. The pipette solution contained: 120
Using a highly sensitive and quantitative chromatographic
mM KCl, 20 mM KOH, 4 mM MgCl2, 5 mM N-
tris (hydroxymethyl)
method, we demonstrated that
fat-3 deletion mutants are
methyl-2-aminoethane-sulphonic acid, 0.25 mM CaCl2, 4 mM
defective in LC-PUFA production and display a fatty acid
NaATP, 36 mM sucrose, 5 mM EGTA, pH 7.2, sucrose to 335 mOsm.
composition similar to that reported for
fat-3(wa22) mutants
All data analysis and graph preparation was performed using Pulsefit
(Watts and Browse, 2002). Wild-type worms produce
(HEKA), Mini Analysis (Synaptosoft, Decatur, GA), and Igor Pro
significant levels of dihomo-γ-linolenic acid (DGLA), AA and
(Wavemetrics, Lake Oswego, OR).
eicosapentaenoic acid (EPA; Table 1), three LC-PUFAssynthesized only in the presence of active ∆6-desaturase.
Consistent with loss of ∆6-desaturase activity,
fat-3(lg8101)
mutant animals have drastically reduced levels of these three
Generation of mutants depleted of LC-PUFAs
LC-PUFAs and accumulate two ∆6-desaturase substrates, ALA
LC-PUFAs are synthesized from dietary precursors by
and LIN (Table 1). The residual levels of DGLA, AA and EPA
sequential double bond insertion (desaturation) and elongation.
detected in these animals may reflect the activity of another
∆6-desaturase catalyses desaturation at the ∆6 position of
desaturase or a dietary contribution to the animal. These
α-linolenic acid (ALA) and LIN (Los and Murata, 1998;
observations suggest that the defects observed in
fat-3 animals
Nakamura et al., 2001) (Fig. 1A). The
C. elegans gene
fat-3,
are caused by depletion of LC-PUFAs.
also called W08D2.4 (Fig. 1B), is a ∆6-desaturase. This proteincontains three histidine clusters distinctive of desaturases(Los and Murata, 1998), harbors a cytochrome-b
The behavioral defects observed in fat-3 mutants are
observed in other ∆6-desaturases (Napier et al., 1999), is
caused by LC-PUFA depletion
homologous to human and plant ∆6-desaturases (Fig. 1C), and
fat-3 mutants display a variety of phenotypes that include both
has ∆6-desaturase enzymatic activity on C18 fatty acids
behavioral and non-behavioral defects (Watts et al., 2003).
(Napier et al., 1998).
Since we were interested in clarifying the role of LC-PUFAs
fat-3(wa22) results in a serine to phenylalanine substitution
in the nervous system, we focused our analysis on the two most
at position 186 (Fig. 1B,C) (Watts and Browse, 2002); this
prominent
fat-3 behavioral phenotypes, namely, the deficits in
serine is not conserved in human or plant desaturases and it is
movement and egg laying.
fat-3 mutants show deficiencies in
not clear that this mutation is a null allele. To determine the
both forward and backward movements, and are particularly
null phenotype we generated two
fat-3 deletion mutations
unable to respond to head-touch, that is, when touched gently
using PCR to screen chemically mutagenized
C. elegans
near the head, wild-type animals respond by reversing direction
libraries with
fat-3 specific primers (Jansen et al., 1997). Both
and proceeding rapidly away from the stimulus. However,
of these mutations are likely to strongly reduce or completely
when
fat-3 mutants are stimulated in the same manner, they
eliminate
fat-3 activity:
fat-3(qa1811) lacks 1,324 bp that
stop or proceed backwards only very slowly. In addition, while
include the three histidine clusters necessary for desaturase
fat-3 mutants do lay eggs, they frequently retain eggs in the
activity (Los and Murata, 1998).
fat-3(lg8101) lacks 2,076 bp
uterus abnormally as they age. Some of these eggs hatch before
that include the start codon, an invariant heme-binding site
being laid, causing the mother to eventually be consumed by
indispensable for enzymatic activity (Sayanova et al., 1999)
hatched embryos. This phenotype is both qualitatively and
and a large portion of the predicted promoter (Fig. 1B) and
quantitatively similar to the egg-laying defects caused by
results in no detectable
fat-3 mRNA transcripts (Fig. 2B,C).
mutations in the
egl-1 gene, which cause a specific
fat-3(lg8101) homozygous animals develop very slowly and
developmental disruption of the hermaphrodite specific
their phenotype is more severe than that of
fat-3(lg8101)/fat-
neurons (HSNs) (Desai and Horvitz, 1989; Trent et al., 1983),
a pair of serotonergic neurons innervating the egg-laying
homozygotes (data not shown). Therefore, it is likely that
fat-
muscles. In particular, 29% of
fat-3(lg8101)/fat-3(qa1811)
3(lg8101) is a molecular null while
fat-3(qa1811) and
fat-
(
n=24), 39% of
fat-3(lg8101) (
n=23) and 47% of
egl-1(n487)
3(wa22) are severe loss-of-function mutations. The phenotype
(
n=34) animals were consumed by hatched embryos late in
of
fat-3(qa1811) homozygous animals could not be directly
adult life. Based on these observations and on the fact that these
assessed because the
qa1811 allele is tightly associated with
behavioral defects were rescued by selective expression of
fat-
an independent lethal mutation.
3 in the nervous system (see below) we hypothesized that
fat-
Table 1. LC-PUFA composition
Percentage (w/w) of total fatty acids
LC-PUFA composition of total phospholipids isolated from wild-type and
fat-3(lg8101) C. elegans grown in normal growth medium or medium containing
DHA. Transgenic
fat-3(lg8101)Exfat-3(+) worms carry cosmid C24G5, which contains coding region and regulatory sequences of
fat-3. Data are themean±s.e.m. of 3-4 independent measurements. ND, not detected (<0.2%).
LC-PUFAs and neurotransmission
Fig. 3. Rescue of the behavioral
defects associated with loss of
fat-3 activity. (A) ExogenousAA and DHA, but not LIN,
rescue the movement defects of
mutants. Animals were exposed
to fatty acids from egg to adult.
*
P<0.0001 versus wild-type
Exfat-3(+) Exfat-3(+) Exfat-3(+)
animals. (B)
fat-3 expressedunder the control of theneuronal promoter
unc-119 (
Exfat-3(+) neuron)
but not under the control of the muscularpromoter
myo-3 (
Exfat-3(+) muscle),
completely rescues the egg-laying defect of
fat-3(lg8101)/fat-3(qa1811) animals. Egl+ indicateshermaphrodites that were not consumed byembryos by the fourth day after reachingadulthood. (C)
fat-3 expressed under the control
of the neuronal promoter but not under the
control of the muscular promoter or theintestinal promoter
elt-2 (
Exfat-3(+) intestine),almost completely rescues the movement
defects of
fat-3 (lg8101) homozygous animals.
*
P<0.0001 versus
fat-3Exfat-3(+) animals. Data
Exfat-3(+) Exfat-3(+)
in A and C are plotted as mean ± s.e.m.
3 activity might be required for normal neuronal development
involved in a variety of biological functions. However,
or function.
eicosanoids do not appear to be produced or used in
C.
To determine whether the behavioral defects observed in
fat-
elegans. Thus, AA exerts its function via a distinct as yet
3 mutants are caused by deficits in LC-PUFA levels, we asked
uncharacterized lipid pathway. Conversion of AA into DHA
whether exogenous LC-PUFAs rescue the movement defects
precursors by a
C. elegans enzyme has been previously
associated with loss of
fat-3 function. We grew
fat-
observed (Spychalla et al., 1997). Therefore, the active LC-
3(lg8101)/fat-3(qa1811) hermaphrodites from egg to adult in
PUFA species are DHA or its related metabolic products and
the presence of AA, whose synthesis is dependent on ∆6-
precursors, which could be generated catabolically.
desaturase (Fig. 1A). AA fully rescued the lack of coordinationof
fat-3 mutants (Fig. 3A). Similarly, exogenous application ofDHA, another LC-PUFA product of ∆6-desaturase activity,
fat-3 expressed in neurons rescues the behavioral
was also sufficient to rescue the locomotion defects associated
defects of fat-3 mutants
with
fat-3 mutant animals. As a negative control we used LIN
The FAT-3 protein is expressed in the intestine, body-wall
(Fig. 1A). Since
fat-3 mutants have inactive ∆6-desaturase and
muscles, pharynx and several neurons (Watts et al., 2003). To
can convert LIN to LC-PUFAs only very poorly, if at all (Table
determine where
fat-3 expression is required, we generated
1), LIN administration is not expected to rescue the
fat-3
constructs that drive gene expression in specific tissues and
phenotype. Indeed, exogenous LIN did not have any effect on
tested their ability to rescue the behavioral defects associated
the motility of
fat-3 mutants (Fig. 3A).
with loss of
fat-3 activity. As expected, a construct comprisingthe endogenous promoter, the coding sequence and the 3′untranslated region of
fat-3 (Fig. 1B) restored normal LC-
DHA or its metabolic products mediate ∆6-desaturase
PUFA levels (Table 1) and rescued the egg-laying impairment
and the reduced motility of
fat-3(lg8101) homozygous animals
To test whether ∆6-desaturase function is mediated by DHA or
(Fig. 3B,C). Since the uncoordinated and egg-laying
AA, we measured the levels of each lipid in
fat-3 mutant
phenotypes of
fat-3 mutants are suggestive of neuronal defects,
animals rescued with DHA. We observed that exogenous
we investigated whether
fat-3 selectively expressed in the
application of DHA did not increase AA levels in
fat-3 mutant
nervous system could rescue the impaired motility and the egg-
animals (Table 1), suggesting that AA by itself is not
laying defect of
fat-3 mutant animals. We made a chimeric
responsible for the observed phenotypic rescue. In other
construct,
Punc-119::fat-3, in which the
fat-3 coding sequence
systems, AA is a precursor of eicosanoids, bioactive lipids
is placed under the control of a promoter driving gene
Journal of Cell Science 116 (24)
expression in the entire nervous system (Maduro and Pilgrim,
serotonergic neurons (Fig. 4B). Moreover, when we visualized
1995). Transgenic
fat-3 mutants bearing this construct
the entire nervous system using a pan-neuronal GFP marker
recovered normal egg-laying capability (Fig. 3B) and almost
(
Punc-119::gfp) we were also unable to detect any
normal motility (Fig. 3C). Conversely, expression of the
fat-3
morphological defect in
fat-3 mutant animals (data not shown).
coding sequence under the control of the muscle-specific
myo-
Finally, when we analyzed the ultrastructure of the nervous
3 promoter did not rescue the egg-laying defect (Fig. 3B) and
system, we found that the general arrangement, structure and
only minimally rescued the motility defect (Fig. 3C) associated
positioning of neurons as well as synaptic morphology are
with the
fat-3 mutation. We also tested whether intestine-
normal in
fat-3 mutants (data not shown). These results suggest
specific expression was sufficient to rescue the
fat-3 mutant
that the neuronal impairments associated with loss of
fat-3
phenotype. We placed
fat-3 under the control of the intestine-
activity are likely to be functional rather than developmental.
specific promoter
elt-2 (Fukushige et al., 1998), which is
If the behavioral defects observed in
fat-3 mutants are indeed
expressed in the intestine and its precursors cells from early
functional it should be possible to rescue these phenotypes by
embryonic stages. This chimeric construct could not rescue the
providing adult animals, in which the nervous system is fully
uncoordinated phenotype of
fat-3 mutants (Fig. 3C). Since we
developed, with the metabolic products of
fat-3 activity. We
restored normal egg-laying and near-normal motility only
therefore exposed adult
fat-3 mutants to exogenous LC-PUFAs
when we expressed
fat-3 in the nervous system, it is likely that
and analyzed movement. Adult homozygotes exposed to AA
fat-3 activity is required in neurons for their normal function.
for 24 hours recovered almost normal motility and were
However, we cannot exclude the possibility that
fat-3 may have
completely rescued after 48 hours (Fig. 4C). These results are
additional normal functions in other cells.
consistent with the notion that LC-PUFAs are required forneuronal function rather than development.
Loss of fat-3 activity causes functional, notdevelopmental defects
fat-3 mutants display defects in neurotransmitter release
The behavioral deficits observed in
fat-3 mutants could reflect
To better assess these defects in neuronal function, we
developmental defects in the nervous system. As a first
measured the transmission efficiency of both serotonergic
approach to this issue, we analyzed the neuronal morphology
NMJs involved in egg-laying and cholinergic NMJs involved
of
fat-3 mutants at the light microscope level. We generated
in body wall muscle contraction and movement. Egg-laying is
animals in which specific subsets of neurons were fluorescently
mainly controlled by the serotonergic HSN motor neurons
labeled with green fluorescent protein (GFP) and could not
(Trent et al., 1983). Both exogenous serotonin and fluoxetine
detect any morphological defect in HSN structure or in the
induce wild-type animals to lay eggs (Desai and Horvitz, 1989;
attachments of the HSN to its egg-laying muscle target (Fig.
Trent et al., 1983; Weinshenker et al., 1995). Fluoxetine
4A). We also could not detect morphological alterations in
potentiates the effect of endogenous serotonin by selectivelyinhibiting its presynaptic re-uptake (Hyttel, 1994). Animalsmissing the HSN neurons are sensitive to serotonin andinsensitive to fluoxetine (Trent et al., 1983; Weinshenker et al.,1995). Animals with defective egg-laying muscles areinsensitive to both drugs. The egg-laying response of
fat-3
mutant animals exposed to serotonin wasnormal (Fig. 5A). For example, at 5mg/ml, the dose eliciting the highestresponse, wild-type animals laid16.42±1.17 eggs and
fat-3(lg8101)/fat-3(qa1811) mutants laid 13.00±1.04 eggs.
This demonstrates that muscle functionin these animals is not disrupted.
However,
fat-3 animals responded very
poorly to fluoxetine (Fig. 5B). Wild-typeanimals laid an increasing number ofeggs when exposed to higher doses offluoxetine. They reached a peak of11.88±0.97 eggs laid at 0.5 mg/mlfluoxetine. However,
fat-3(lg8101)/fat-3(qa1811)
animals laid an almost
constant number of eggs whatever the
Fig. 4. fat-3 mutant animals display functional and not morphological neuronal defects.
dose of fluoxetine (from 3.29±0.64 eggs
(A,B) Serotonergic neurons visualized with GFP under the control of a tryptophane
at 0.1 mg/ml fluoxetine to 5.00±0.94
hydoxylase promoter (
tph-1), which is expressed in serotonergic neurons (Sze et al., 2000).
eggs at 1 mg/ml). Similarly,
fat-3
(A) An HSN motor neuron in proximity of the vulva. Bars, 5 µm. (B) Serotonergic neurons
mutants were only inefficiently
in the head. Images shown are projections of confocal xy sections. Bars, 10 µm.
stimulated by imipramine, another
(C) Exogenous arachidonic acid (AA) restores wild-type movement in adult
fat-3(lg8101)/fat-3(qa1811) mutant animals within 48 hours. *
P=0.0017 versus wild-type
potentiator of endogenous serotonin
animals. Data is plotted as mean ± s.e.m.
(Fig. 5C). These observations suggest
LC-PUFAs and neurotransmission
Levamisole after AA
fat-3Exfat-3(+) neuron
fat-3Exfat-3(+) muscle
Fig. 5. Cholinergic and serotonergic synapses are functionally disrupted in
fat-3 mutant animals. (A-C) Synaptic release of endogenous
serotonin was measured by determining the egg-laying response of
fat-3(lg8101)/fat-3(qa1811) mutants to serotonin, fluoxetine and
imipramine. Muscles are functional in
fat-3 mutants because they respond well to serotonin (A,C). However, they are defective in serotonin
release, because they respond only inefficiently to the endogenous serotonin potentiators fluoxetine (B) and imipramine (C). In C, a single dose
of serotonin (5 mg/ml) and imipramine (0.75 mg/ml) were used. (D,E) Synaptic release of endogenous ACh was measured by determining the
response to the endogenous ACh potentiator aldicarb.
fat-3 mutants are defective in ACh release because aldicarb induces spastic paralysis
faster in wild-type animals than in
fat-3(lg8101)/fat-3(qa1811) (D) or in
fat-3(wa22) (E) mutants. The defect of
fat-3 mutants is weaker but
comparable to that of
rab-3(y250), a synaptic vesicle transmission mutant. *
P<0.01; **
P<0.005. (F) The altered aldicarb sensitivity of
fat-3
mutants is of neuronal origin.
fat-3 expressed in neurons, but not in muscles, restores normal aldicarb sensitivity to
fat-3(lg8101) mutants. (G-
H) The ACh mimetic levamisole induces paralysis faster in
fat-3(lg8101)/fat-3(qa1811) (G) and in
fat-3(wa22) (H) mutants than in wild-type
animals. *
P<0.003. (I) Normal sensitivity to levamisole is restored in
fat-3(lg8101)/fat-3(qa1811) mutant animals exposed to AA from egg to
adult. In all panels, data points show the mean ± s.e.m.
that
fat-3 mutant animals release abnormally low levels of
paralysis. However, mutants with decreased ACh release are
serotonin into the synaptic cleft.
resistant to aldicarb (Jorgensen et al., 1995; Nonet et al., 1993;
Cholinergic NMJ function was probed using the agonist
Nonet et al., 1998). Consistent with reduced ACh release into
levamisole, which binds to muscular ACh receptors, and the
the synaptic cleft,
fat-3 mutant animals were significantly less
ACh esterase inhibitor aldicarb, which enhances the effect of
responsive to aldicarb than wild-type animals. For example,
endogenously released ACh. Treatment of wild-type animals
exposure to 1 mM aldicarb for 60 minutes resulted in
with either drug results in muscle hypercontraction and
90.5±3.3% of wild-type animals and only 54.5±7.4% of
fat-
Journal of Cell Science 116 (24)
3(lg8101)/fat-3(qa1811) mutant animals being paralyzed (Fig.
defects in ACh biosynthesis are unlikely to account for the
5D). Both
fat-3(lg8101)/fat-3(qa1811)
and
fat-3(wa22)
movement defects displayed by
fat-3 mutants. Taken together,
animals responded to aldicarb in similar fashion (Fig. 5D,E).
these results suggest that the
fat-3 lesion causes inefficient ACh
This reduced response to aldicarb is comparable in severity to
release from cholinergic neurons.
that observed in the "weak" synaptic transmission mutant
rab-
To test directly whether
fat-3 mutants have decreases in ACh
3 (Fig. 5D) (Nonet et al., 1997). To verify that this response is
release, we measured both evoked excitatory postsynaptic
associated with loss of
fat-3 activity, we exposed
fat-3 mutants,
currents and endogenous miniature excitatory postsynaptic
expressing
fat-3 in neurons, to aldicarb. These, but not animals
currents from voltage-clamped muscles at the
C. elegans NMJ.
expressing
fat-3 in muscles, recovered almost normal
fat-3(wa22) mutants displayed a decrease in evoked amplitude
sensitivity to aldicarb (Fig. 5F). This decreased response is not
(417±41 pA) compared to the wild type (830±144 pA; Fig.
due to ACh receptor impairment because
fat-3 mutants were
6A,B). In addition, the frequency of miniature postsynaptic
hypersensitive to both levamisole (Fig. 5G,H) and ACh (data
currents, caused by the fusion of one or a few synaptic vesicles,
not shown) as compared to wild-type animals. For example,
was reduced in
fat-3(wa22) mutants (10±2 fusions/second)
exposure to levamisole for 40 minutes resulted in paralysis of
compared to the wild type (15±1 fusions/second; Fig. 6C,D).
47.5±7.1% of wild-type and 94.7±1.5% of
fat-3(lg8101)/fat-
The amplitude of the miniature currents was not significantly
3(qa1811) mutant animals (Fig. 5G). Normal response to
different in
fat-3(wa22) (41±3 pA) compared to wild type
levamisole was restored in
fat-3 mutants grown in the presence
(37±2 pA; Fig. 6E). These data suggest that in
fat-3 mutants
of AA (Fig. 5I), confirming that the hypersensitivity to
synaptic vesicles are correctly filled with neurotransmitter and
levamisole is dependent upon LC-PUFA levels. Such
the postsynaptic receptor field is normal. However, synaptic
hypersensitivity may reflect an adaptive response to decreased
vesicles are either reduced in number or in release probability
ACh availability and is observed in other mutant backgrounds
at
fat-3 mutant synapses.
defective in cholinergic transmission (Nonet et al., 1993).
Lower levels of ACh at the synaptic cleft of
fat-3 mutants couldarise from either inefficient release of ACh or decreased ACh
Presynaptic sites are depleted of synaptic vesicles in fat-
biosynthesis. To discriminate between these possibilities we
quantitatively assessed the level of ACh produced in
fat-3
Decrease in neurotransmitter release in
fat-3 mutants could be
mutants. Since total ACh levels do not diminish in
fat-3
caused by a number of possible mechanisms. For example,
mutants [65.1±10.8 fmoles per µg of protein in
fat-3 mutants
synaptic vesicles could be assembled and localized normally to
(
n=20) versus 48.7±4.0 in wild-type (
n=20),
P=0.3040],
NMJs but undergo exocytosis only inefficiently. In this view,
the number of vesicles at NMJs is expected toincrease. Alternatively,
fat-3 mutants couldlocalize fewer synaptic vesicles at the nerveterminal or could be defective in endocytosis.
In this view, the number of synaptic vesicles isexpected to decrease (Harris et al., 2000;Jorgensen et al., 1995). To distinguish betweenthese possibilities, we examined the synapticultrastructure of
fat-3 mutants and determinedthe distribution of synaptic vesicles at NMJs.
We found that while synapses in wild-typeanimals had clusters of vesicles in theproximity of the active zone (Fig. 7A),synapses in
fat-3 mutants were depleted ofvesicles (Fig. 7B). The number of synapticvesicles per synaptic terminal (within 300 nmof the active zone) was 2.7-fold smaller in
fat-3 mutants than in wild-type animals(10.32±0.96 versus 28.07±3.22,
P<0.0001;Fig. 7C). In addition, both the number ofsynaptic vesicles docked to the presynapticmembrane (0.72±0.15 versus 2.00±0.36,
P<0.001) and the number of those in itsvicinity (within 100 nm; 3.40±0.30 versus6.88±0.87,
P=0.0001) were significantly
Fig. 6. fat-3 mutants have reduced evoked amplitude and reduced rates of spontaneous
decreased in
fat-3 mutant animals (Fig. 7C).
fusion but normal quantal size. (A) Representative evoked responses from wild-type
Moreover, our ultrastructural analysis did not
and
fat-3 animals. (B) The mean amplitude of the evoked responses is reduced in
fat-3
reveal accumulation of vesicles in neuronal
(
n=6) compared to wild-type (
n=7) animals (
P<0.03). (C) Representative traces of
cell bodies in
fat-3 mutants (data not shown).
spontaneous fusion events in wild-type and
fat-3 mutants. (D-E) The mean frequency of
These results indicate that LC-PUFAs are
spontaneous fusion is reduced in
fat-3 mutants (
n=13) compared to wild-type (
n=14;*
P<0.02), while the mean amplitude of the individual events is normal. Data is plotted
required to maintain a normal pool of synaptic
as mean ± s.e.m.
vesicles at NMJs.
LC-PUFAs and neurotransmission
PUFAs causes functional rather than developmental defects inthe nervous system. These defects can be rescued by selectiveFAT-3 expression in the nervous system or by dietarysupplementation of LC-PUFAs to adult worms. Usingpharmacological techniques, we identify neurotransmitterrelease defects in both cholinergic and serotonergic neuronsof fat-3 mutants. Electrophysiological studies suggest thata
decrease in neurotransmitter release rather than
neurotransmitter loading is responsible for the neuronal defectsof fat-3 mutants. Finally, ultrastructural analysis of synapticterminals demonstrates that synapses are depleted of synapticvesicles. We conclude that LC-PUFA depletion results ininsufficient neurotransmitter release.
LC-PUFAs and neurotransmitter releaseThe locomotion and egg-laying defects observed in fat-3mutants are neuronal in nature and could be rescued byexpressing fat-3 in the nervous system but not by expressing itin muscles or intestine. This suggests that LC-PUFAs areproduced and act in neurons. The defects associated with lossof fat-3 activity were also rescued by providing exogenous LC-PUFAs. Free fatty acids are known to diffuse across biologicalmembranes from intercellular spaces (Frohnert and Bernlohr,2000). The observation that fat-3 expressed in intestine andmuscles did not result in rescue, suggests that fat-3 expressionin these tissues does not provide enough LC-PUFAs inintercellular spaces to support normal neuronal function.
Therefore it is likely that fat-3 activity is required in neuronsfor their normal function.
Although we cannot rule out that fat-3 mutants might have
subtle neuronal developmental defects, several lines ofevidence suggest that the neuronal developmental defects donot contribute significantly to the behavioral phenotypesobserved in C. elegans depleted of LC-PUFAs. First, in fat-3mutants we could not observe gross morphological defectsin neurons visualized with GFP markers. Second, at theultrastructural level we found that both the general organizationof the nervous system and neuronal specializations such as theNMJs appeared normal in animals without fat-3 activity. Third,the locomotion defects of adult fat-3 mutants were rescuedacutely by providing exogenous LC-PUFAs. Therefore, our
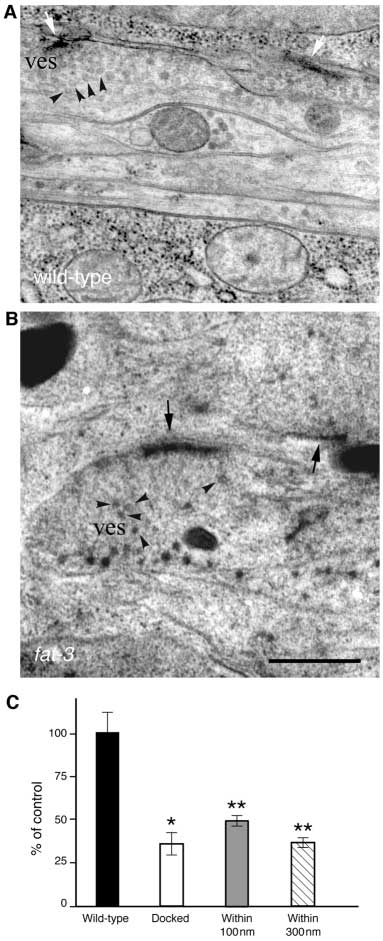
Fig. 7. Synapses are partially depleted of synaptic vesicles in fat-3
results indicate that LC-PUFAs are important for neuronal
mutant animals. Electron micrographs of wild-type (A) and fat-
function rather than neuronal development.
3(lg8101) (B) synapses fixed by fast-freezing. (A) Clusters of
Two lines of evidence indicate the defects in neuronal
vesicles (ves and arrowheads) in ventral nerve cord neurons arelocalized close or docked to the active zone (white arrows).
function observed in fat-3 mutants are due to decreases in
(B) Ventral ganglion synapses are depleted of most vesicles close to
neurotransmitter release. First, fat-3 mutant animals displayed
the active zone (black arrows). A few vesicles lie at a distance (ves
presynaptic defects in neuronal function at both cholinergic
and arrowheads). (C) Quantification of synaptic vesicles in fat-
and serotonergic neurons in pharmacological assays. Second,
3(lg8101)/fat-3(qa1811) mutant animals. The average fraction of
electrophysiological recordings demonstrate that fat-3 mutants
synaptic vesicles per section at given distances in fat-3 mutants
release abnormally low levels of neurotransmitter. This
compared to wild-type animals (100%). Vesicles were considered
impaired neurotransmission is probably caused by decreases in
docked when they were touching the active zone. Total number of
synaptic vesicle number rather than neurotransmitter loading
synapses scored: 50, fat-3; 42, wild-type. *P<0.001; **P<0.0001.
or release probability. In fat-3 mutants an abnormally low
Scale bar: 0.5 µm.
quantity of neurotransmitter is released upon stimulation. Thisreduction in neurotransmitter release does not result from
decreases in loading of neurotransmitter into synaptic vesicles
The complex lipids LC-PUFAs are highly enriched and
as fat-3 mutants have similar amplitudes of mini currents as
precisely regulated in neurons. By inactivating the gene fat-3,
wild-type animals. Thus, a decrease in the number of synaptic
we have generated animals depleted of LC-PUFAs and
vesicles undergoing exocytosis is probably responsible for the
analyzed their neuronal deficits. We show that depletion of LC-
impaired neurotransmission. This reduced number of vesicles
Journal of Cell Science 116 (24)
undergoing exocytosis is probably due to a decrease in
1988). Conversely, Weisinger and collegues reported a
available vesicles rather than defects in release since our
complete functional recovery (Weisinger et al., 1999). Here we
ultrastructural analysis of fat-3 mutant synapses showed
have demonstrated that LC-PUFA depletion leads to functional
reductions in both total and morphologically docked synaptic
but not developmental defects in C. elegans neurons. Our
vesicles. The decrease in available vesicles being responsible
findings are consistent with recent reports showing that
for fat-3 mutant defects is also supported by the correlation
supplementation of certain LC-PUFAs can improve
between the extent of synaptic vesicle depletion and the
neurological conditions associated with LC-PUFA mis-
decrease in synaptic vesicle fusion. fat-3 mutants carry
regulation (Martinez et al., 2000).
approximately 40% of the synaptic vesicles of wild-type
These experiments provide a foundation for a genetic
animals. Similarly, the evoked responses in fat-3 mutants is
analysis of LC-PUFA function. We anticipate that genetic
approximately 50% that of wild-type animals. We conclude
screens aimed at identifying proteins regulated by LC-PUFAs
that the abnormally low number of synaptic vesicles present at
in C. elegans will uncover the cellular targets and define the
NMJs of fat-3 mutants is insufficient to support normal
molecular mechanisms underlying the roles of LC-PUFAs in
neurotransmission.
The decrease in synaptic vesicle number at synaptic
terminals could be due to defects in transport, endocytosis, or
This work was supported by the Wellcome Trust (G.M.L.), by
synaptic vesicle biogenesis. fat-3 mutants are unlikely to have
Cancer Research UK (G.M.L. and G.S.), by The Natural Sciences and
significant defects in synaptic vesicle transport, since in
Engineering Research Council of Canada (M.T.C.) and by the NIH,grant RR12596 (D.H.H.). We thank Tom R. Clandinin, Neil Hopper,
our ultrastructural analysis we did not observe vesicle
Enzo Lalli, Raffi Aroian, Cori Bargmann, Amanda Kahn-Kirby, Julian
accumulation in the proximity of the Golgi apparatus or in the
Lewis, Caetano Reis e Sousa, Nirao Shah, Jennifer Watts, Axel
rest of the neuronal cell body (data not shown). Therefore it is
Behrens and Ralf Adams for critically reading the manuscript, Tom
likely that fat-3 mutants are defective in synaptic vesicle
R. Clandinin for advice and discussions, Yeow Goh for fatty acid
biogenesis and/or synaptic vesicle recycling. Further work is
analysis, Ji Ying Sze and Gary Ruvkun for strains, John Sulston for
needed to clarify in detail the mechanism(s) responsible for the
cosmids, Andy Fire for plasmids, Giovanna Lalli for help with
synaptic vesicle depletion observed in fat-3 mutant animals.
confocal microscopy, Graham Clark for help with PCR screening, andKen Nguyen, Toni Long and Nancy Hall for help with EM analysis.
Some nematode strains were supplied by the Caenorhabditis Genetics
Link with human brain function
Center, which is supported by the NIH and the University ofMinnesota.
LC-PUFAs have been associated with normal neuronal andretinal function (Lauritzen et al., 2001; Martinez et al., 2000;Meloni et al., 2002). In addition, it has been suggested that AA
and DHA are required for normal cognitive development
Anderson, J. W., Johnstone, B. M. and Remley, D. T. (1999). Breast-feeding
in infants (e.g. Anderson et al., 1999; Helland et al., 2003;
and cognitive development: a meta-analysis. Am. J. Clin. Nutr. 70, 525-535.
Willatts et al., 1998). This hypothesis derives from studies
Brenner, S. (1974). The genetics of Caenorhabditis elegans. Genetics 77, 71-
comparing cognitive function of breast-fed infants with those
of formula-fed infants. In most of these studies children who
Chomczynski, P. and Sacchi, N. (1987). Single-step method of RNA isolation
by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal.
had been breast-fed scored better than children who had been
Biochem. 162, 156-159.
formula fed. Since human milk naturally contains AA and
Connor, W. E. and Neuringer, M. (1988). The effects of n-3 fatty acid
DHA, while infant formulas do not, it was concluded that AA
deficiency and repletion upon the fatty acid composition and function of the
and DHA are responsible for the better cognitive skills
brain and retina. Prog. Clin. Biol. Res. 282, 275-294.
Desai, C. and Horvitz, H. R. (1989). Caenorhabditis elegans mutants
observed in breast-fed infants. However, it has been difficult to
defective in the functioning of the motor neurons responsible for egg laying.
unequivocally determine whether this difference is due to LC-
Genetics 121, 703-721.
PUFAs or to confounding variables such as socio-economic
Frohnert, B. I. and Bernlohr, D. A. (2000). Regulation of fatty acid
status or parental education. Moreover, no specific cellular
transporters in mammalian cells. Prog. Lipid Res. 39, 83-107.
process has been unambiguously identified for LC-PUFAs.
Fukushige, T., Hawkins, M. G. and McGhee, J. D. (1998). The GATA-factor
elt-2 is essential for formation of the Caenorhabditis elegans intestine. Dev.
Here we have demonstrated pharmacologically and
Biol. 198, 286-302.
electrophysiologically that LC-PUFA depletion leads to
Hall, D. H. (1995). Electron microscopy and three-dimensional image
decreased neurotransmitter release and have therefore proved
reconstruction. Methods Cell Biol. 48, 395-436.
that LC-PUFAs have a direct effect on neuronal function. Thus
Hargreaves, K. M. and Clandinin, M. T. (1988). Dietary control of
diacylphosphatidylethanolamine species in brain. Biochim. Biophys. Acta
LC-PUFA depletion is likely to impair communication among
962, 98-104.
many types of neurons. This could well have a dramatic effect
Harris, T. W., Hartwieg, E., Horvitz, H. R. and Jorgensen, E. M. (2000).
on cognition and memory and could account for the
Mutations in synaptojanin disrupt synaptic vesicle recycling. J. Cell Biol.
psychomotor retardation associated with diseases altering LC-
PUFA metabolism (Martinez et al., 2000; Meloni et al., 2002).
Helland, I. B., Smith, L., Saarem, K., Saugstad, O. D. and Drevon, C. A.
(2003). Maternal supplementation with very-long-chain n-3 fatty acids
It is still controversial as to whether LC-PUFA deficiency
during pregnancy and lactation augments children's IQ at 4 years of age.
leads to functional or developmental defects in humans
Pediatrics 111, e39-e44.
(Lauritzen et al., 2001). For example, conflicting data exist on
Hyttel, J. (1994). Pharmacological characterization of selective serotonin
the recovery of retinal function after repletion of LC-PUFA
reuptake inhibitors (SSRIs). Int. Clin. Psychopharmacol. 9 Supplement 1,
19-26.
depleted animals. Connor and Neuringer reported that
Jansen, G., Hazendonk, E., Thijssen, K. L. and Plasterk, R. H. (1997).
restoration of normal LC-PUFA levels was not sufficient to
Reverse genetics by chemical mutagenesis in Caenorhabditis elegans.
rescue functional retinal abnormalities (Connor and Neuringer,
Nature Genet. 17, 119-121.
LC-PUFAs and neurotransmission
Jorgensen, E. M., Hartwieg, E., Schuske, K., Nonet, M. L., Jin, Y. and
Nonet, M. L., Staunton, J. E., Kilgard, M. P., Fergestad, T., Hartwieg, E.,
Horvitz, H. R. (1995). Defective recycling of synaptic vesicles in
Horvitz, H. R., Jorgensen, E. M. and Meyer, B. J. (1997). Caenorhabditis
synaptotagmin mutants of Caenorhabditis elegans. Nature 378, 196-199.
elegans rab-3 mutant synapses exhibit impaired function and are partially
Lackner, M. R., Nurrish, S. J. and Kaplan, J. M. (1999). Facilitation of
depleted of vesicles. J. Neurosci. 17, 8061-8073.
synaptic transmission by EGL-30 Gqα and EGL-8 PLCβ: DAG binding
Richmond, J. E., Davis, W. S. and Jorgensen, E. M. (1999). UNC-13 is
to UNC-13 is required to stimulate acetylcholine release. Neuron 24, 335-
required for synaptic vesicle fusion in C. elegans. Nat. Neurosci. 2, 959-
Lauritzen, L., Hansen, H. S., Jorgensen, M. H. and Michaelsen, K. F.
Richmond, J. E. and Jorgensen, E. M. (1999). One GABA and two
(2001). The essentiality of long chain n-3 fatty acids in relation to
acetylcholine receptors function at the C. elegans neuromuscular junction.
development and function of the brain and retina. Prog. Lipid Res. 40, 1-94.
Nat. Neurosci. 2, 791-797.
Los, D. A. and Murata, N. (1998). Structure and expression of fatty acid
Sayanova, O., Shewry, P. R. and Napier, J. A. (1999). Histidine-41 of the
desaturases. Biochim. Biophys. Acta 1394, 3-15.
cytochrome b5 domain of the borage delta6 fatty acid desaturase is essential
Maduro, M. and Pilgrim, D. (1995). Identification and cloning of unc-119,
for enzyme activity. Plant Physiol. 121, 641-646.
a gene expressed in the Caenorhabditis elegans nervous system. Genetics
Sharon, R., Bar-Joseph, I., Frosch, M. P., Walsh, D. M., Hamilton, J. A.
and Selkoe, D. J. (2003). The formation of highly soluble oligomers of
Martinez, M., Vazquez, E., Garcia-Silva, M. T., Manzanares, J., Bertran,
alpha-synuclein is regulated by fatty acids and enhanced in Parkinson's
J. M., Castello, F. and Mougan, I. (2000). Therapeutic effects of
disease. Neuron 37, 583-595.
docosahexaenoic acid ethyl ester in patients with generalized peroxisomal
Spychalla, J. P., Kinney, A. J. and Browse, J. (1997). Identification of an
disorders. Am. J. Clin. Nutr. 71, 376S-385S.
animal omega-3 fatty acid desaturase by heterologous expression in
McDonald, K. (1999). High-pressure freezing for preservation of high
Arabidopsis. Proc. Natl Acad. Sci. USA 94, 1142-1147.
resolution fine structure and antigenicity for immunolabeling. Methods Mol.
Sze, J. Y., Victor, M., Loer, C., Shi, Y. and Ruvkun, G. (2000). Food and
Biol. 117, 77-97.
metabolic signalling defects in a Caenorhabditis elegans serotonin-
Mello, C. C., Kramer, J. M., Stinchcomb, D. and Ambros, V. (1991).
synthesis mutant. Nature 403, 560-564.
Efficient gene transfer in C. elegans after microinjection of DNA into
Trent, C., Tsung, N. and Horvitz, H. R. (1983). Egg-laying defective mutants
germline cytoplasm: extrachromosomal maintenance and integration of
of the nematode Caenorhabditis elegans. Genetics 104, 619-647.
transforming sequences. EMBO J. 10, 3959-3970.
Wallis, J. G., Watts, J. L. and Browse, J. (2002). Polyunsaturated fatty
Meloni, I., Muscettola, M., Raynaud, M., Longo, I., Bruttini, M., Moizard,
acid synthesis: what will they think of next? Trends Biochem. Sci. 27, 467-
M. P., Gomot, M., Chelly, J., des Portes, V., Fryns, J. P. et al. (2002).
FACL4, encoding fatty acid-CoA ligase 4, is mutated in nonspecific X-
Watts, J. L. and Browse, J. (2002). Genetic dissection of polyunsaturated
linked mental retardation. Nature Genet. 30, 436-440.
fatty acid synthesis in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA
Miller, K. G., Emerson, M. D. and Rand, J. B. (1999). Goα and
diacylglycerol kinase negatively regulate the Gqα pathway in C. elegans.
Watts, J. L., Phillips, E., Griffing, K. R. and Browse, J. (2003). Deficiencies
Neuron 24, 323-333.
in C20 polyunsaturated fatty acids cause behavioral and developmental
Nakamura, M. T., Cho, H. P., Xu, J., Tang, Z. and Clarke, S. D. (2001).
defects in Caenorhabditis elegans fat-3 mutants. Genetics 163, 581-589.
Metabolism and functions of highly unsaturated fatty acids: an update.
Weinshenker, D., Garriga, G. and Thomas, J. H. (1995). Genetic and
Lipids 36, 961-964.
pharmacological analysis of neurotransmitters controlling egg laying in C.
Napier, J. A., Hey, S. J., Lacey, D. J. and Shewry, P. R. (1998). Identification
elegans. J. Neurosci. 15, 6975-6985.
of a Caenorhabditis elegans ∆6-fatty-acid-desaturase by heterologous
Weisinger, H. S., Vingrys, A. J., Bui, B. V. and Sinclair, A. J. (1999). Effects
expression in Saccharomyces cerevisiae. Biochem. J. 330, 611-614.
of dietary n-3 fatty acid deficiency and repletion in the guinea pig retina.
Napier, J. A., Michaelson, L. V. and Stobart, A. K. (1999). Plant desaturases:
Invest. Ophthalmol. Vis. Sci. 40, 327-338.
harvesting the fat of the land. Curr. Opin. Plant Biol. 2, 123-127.
Willatts, P., Forsyth, J. S., DiModugno, M. K., Varma, S. and Colvin, M.
Nonet, M. L., Grundahl, K., Meyer, B. J. and Rand, J. B. (1993). Synaptic
(1998). Effect of long-chain polyunsaturated fatty acids in infant formula on
function is impaired but not eliminated in C. elegans mutants lacking
problem solving at 10 months of age. Lancet 352, 688-691.
synaptotagmin. Cell 73, 1291-1305.
Zhang, K., Kniazeva, M., Han, M., Li, W., Yu, Z., Yang, Z., Li, Y., Metzker,
Nonet, M. L., Saifee, O., Zhao, H., Rand, J. B. and Wei, L. (1998). Synaptic
M. L., Allikmets, R., Zack, D. J. et al. (2001). A 5-bp deletion in ELOVL4
transmission deficits in Caenorhabditis elegans synaptobrevin mutants. J.
is associated with two related forms of autosomal dominant macular
Neurosci. 18, 70-80.
dystrophy. Nature Genet. 27, 89-93.
Source: http://www.zen99974.zen.co.uk/Lab_site/PDF/JCS,%20116,%204965.pdf
ORIGINAL INVESTIGATION HEALTH CARE REFORM An Empirical Model to Estimate the Potential Impactof Medication Safety Alerts on Patient Safety, HealthCare Utilization, and Cost in Ambulatory Care Saul N. Weingart, MD, PhD; Brett Simchowitz, BA; Harper Padolsky, MD; Thomas Isaac, MD, MBA, MPH;Andrew C. Seger, PharmD; Michael Massagli, PhD; Roger B. Davis, ScD; Joel S. Weissman, PhD
Gabinete de Alcaldía Alkatetzako Kabinetea REGLAMENTO ORGÁNICO DEL AYUNTAMIENTO DE PAMPLONA. Pza Consistorial s/n, 3º 31001 Pamplona www.pamplona.es Udaletxe Plaza z/g, 3º 31001 Iruña Gabinete de Alcaldía Alkatetzako Kabinetea El Pleno del excelentísimo Ayuntamiento de Pamplona con fecha 27 de febrero de 1998, aprobó el Reglamento Orgánico del Ayuntamiento de Pamplona.