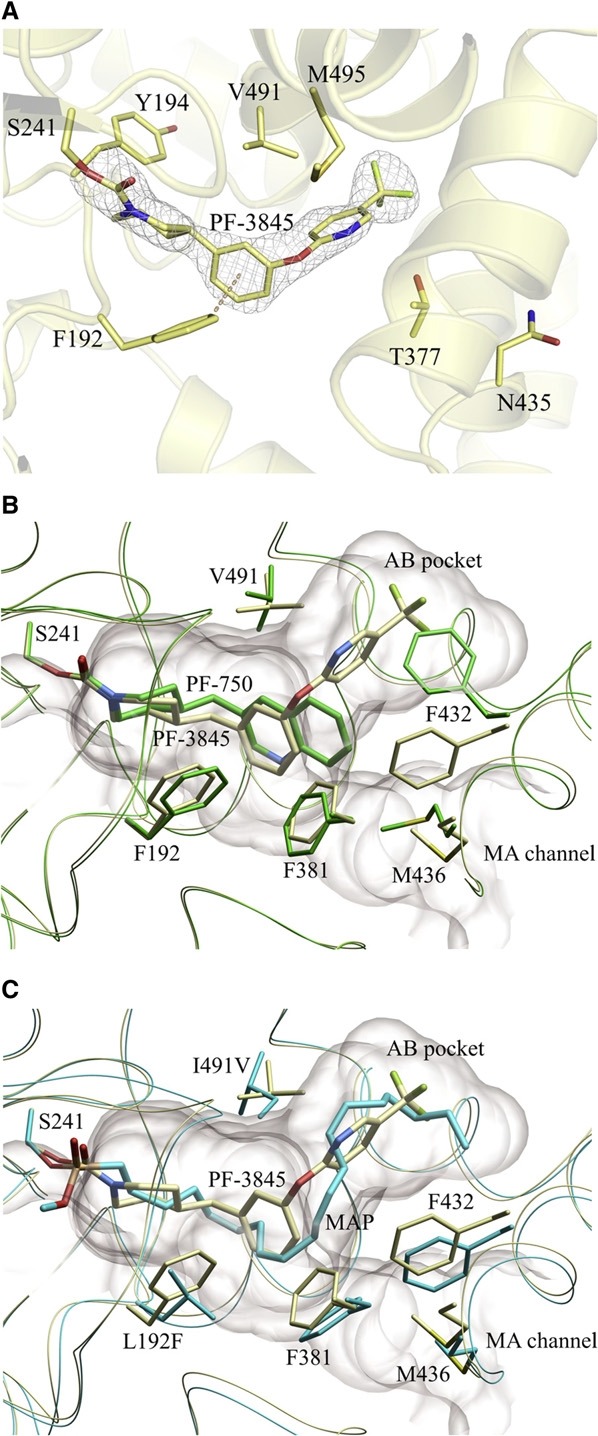
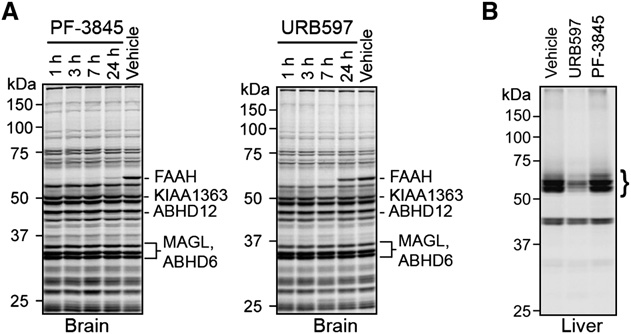
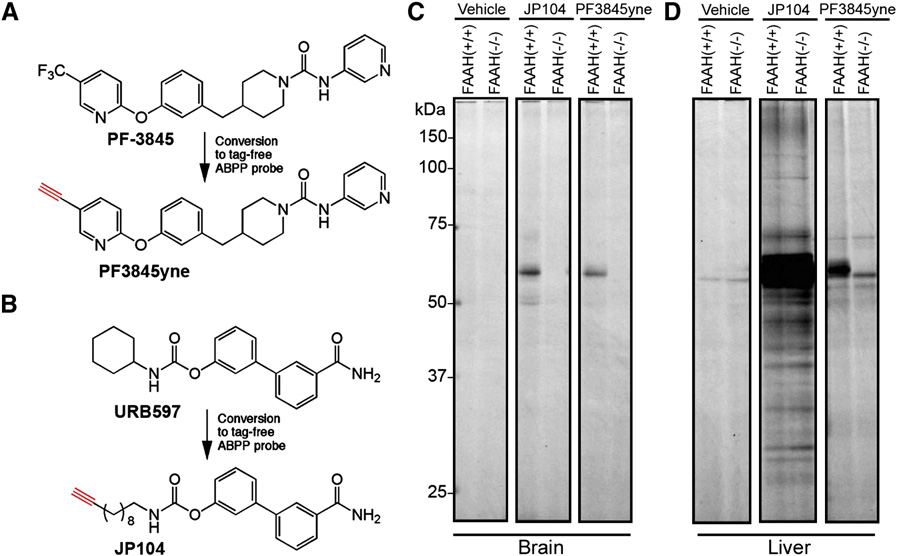
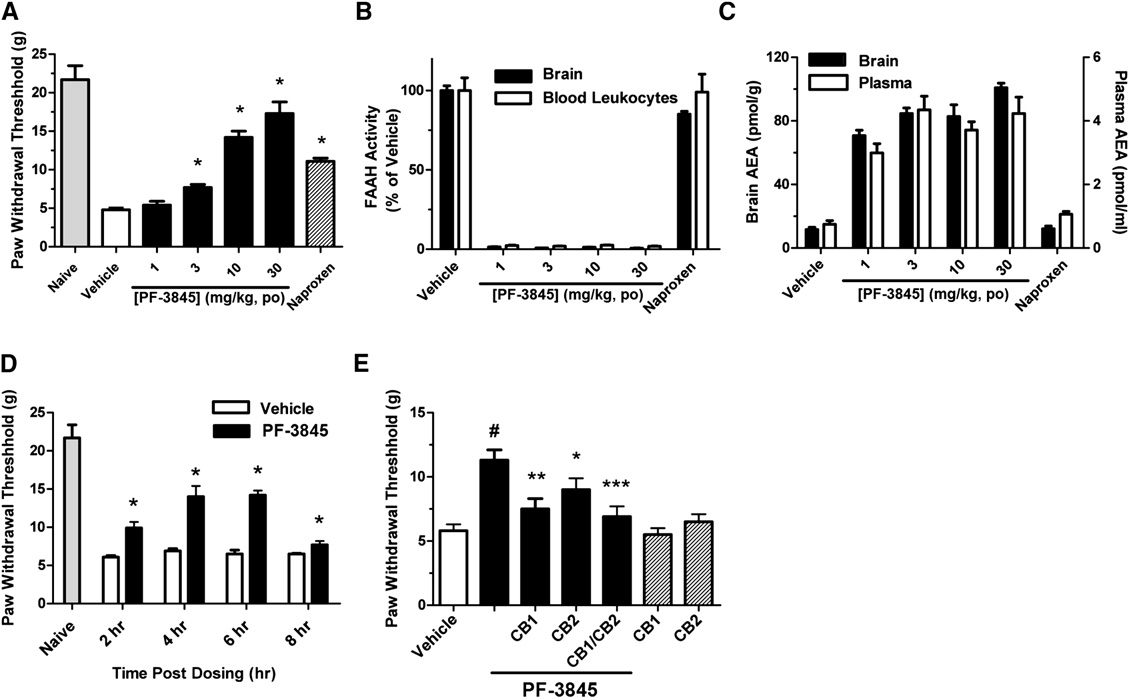
Doi:10.1016/j.hepres.2006.06.01
Hepatology Research xxx (2006) xxx–xxx Short communication Hepatitis B virus infections in families in which the mothers are negative but the fathers are positive for HBsAg Kunio Takegoshi , Wei Zhang Takegoshi Internal Medicine Clinic, 377-7 Nomura, Takaoka, Toyama 933-0014, Japan Department of Pathology, Tangdu Hospital, the Fourth Military Medical University, Xi'an, Shaanxi 710038, China