Sven enterlein
praktikum
Versuch Nummer F-09/10:
Isolierung von Ribosomen
und Bestimmung ribosoma-
Sven Enterlein 108 097 236 174
Versuch Nummer FF---009
I. Einleitung
Die Zentrifugation bildet eine effektive Möglichkeit, Stoffe nach Größe und Gestalt zu trennen. Die Trennung kann entweder einfach zwischen einer festen und einer flüssigen Phase erfolgen, aber auch unterschiedlich große Partikel können (aus einer Lösung) ge-trennt werden. Physikalische Einflüsse dabei sind
Reibung, Auftrieb, Viskosität, Dichte und
Zentrifugalkraft. Die treibende Kraft ist die letztgenannte Zentrifugalkraft; sie steht senkrecht zur Drehachse und ist nach außen gerichtet. Durch sie erfahren die Teilchen eine Beschleunigung, die als Zentrifugalbeschleunigung bezeichnet wird. Sie hängt vom Radius
r und der Winkelgeschwindigkeit ! ab über
z =
r ⋅ !
Da überall auch noch die gewöhnliche Erdbeschleunigung angreift, hat man die Relative Zentrifugalbeschleunigung RCF definiert:
Nach einigen weiteren physikalischen Überlegungen kommt man für den
Sedimentati-onskoeffizienten S eines Teilchens auf folgende Formel:
! ⋅
r ⋅
V ⋅(" − " )
(Die Einheit ist
10− s und wird als SVEDBERG bezeichnet)
Hierbei sind
V das Volumen und " die Dichte des Teilchens, "0 die Dichte des Mediums und
f der Reibungskoeffizient. Unter Einbeziehung des Gesetz von STOKES kann man (über mehrere Zwischenschritte) auf die Molmasse schließen:
RT ⋅ ln
1
r
(Differentialzentrifugation)
D (1 −
v ⋅ " )
(
t2
t1 )
Die Variablen haben folgende Bedeutungen:
(
t − Zeitdifferenz für Strecke ( 2
r −
Diffusionskoeffizient des Teilchens
Versuch Nummer FF---009
Absolute Temperatur × Gaskonstante
Dichte des Mediums
Partielles spezifisches Volumen des Teilchens
Andere Verfahren beruhen auf der Gleichgewichtseinstellung zwischen Sedimentation und diffuser Gleichverteilung. Im Zentrifugenröhrchen herrscht ein Dichtegradient des Mediums (oft Sucrose oder CsCl). Die Berechnung erfolgt dann nach
2 ⋅
RT ⋅ln
(Dichtegradientenzentrifugation)
R = (1 −
v ⋅ " ) 2
Sie sind mit die wichtigsten Bestandteile einer jeden lebenden Zelle, denn mit ihrer Hilfe wird die Basensequenz in eine Aminosäuresequenz übersetzt (
Translation). Es handelt sich dabei um elektronenmikroskopisch sichtbare Teilchen mit ellipsoider Gestalt. Sie können entweder frei im Cytosol vorkommen oder bei Eukaryoten auch gebunden am rauen endoplasmatischen Reticulum (rER). Es gibt zahlreiche Unterschiede in der Translation bei Eukaryoten und Prokaryoten. Wichtig ist natürlich die räumliche Trennung durch den Zellkern. Näher möchte ich darauf nicht eingehen. Erwähnt seien allerdings noch die unterschiedlichen Ribosomen: Bei Prokaryoten haben sie eine Größe von 70 S und bestehen aus einer 50 S- und einer 30 S-Untereinheit. (Anmerkung: Da die Sedimentationsgeschwindigkeit auch von der Gestalt abhängt, verhalten sich die Größen nicht additiv!) Eukaryotische Zellen weisen 80 S Ribosomen auf, die sich aus einer 60 S und einer 40 S Untereinheit zusammen set-zen. Sie werden im Nucleolus des Zellkerns gebildet. Lange Zeit wurde angenommen, dass die katalytische Aktivität auf Proteine zurückzuführen ist. Bald stellte sich aber heraus, dass die enthaltenen Ribonukleinsäuren das katalytische Zentrum bilden. Die kleine 40 S Untereinheit enthält eine 18 S RNA und die große 60 S eine 28 S RNA. Der Aufbau von Nukleinsäuren dürfte wohl bekannt sein. Hier sollen nur kurz die in Ribo-somen mit vorkommenden RNAs charakterisiert werden:
Sedimentationskoeffizient 80S
18S, 1874 Nukleotide
28S, 4178 Nukleotide
5.8S, 160 Nukleotide 5S, 120 Nukleotide
Massenverhältnis 60%
Proteinmasse (kD)
Massenverhältnis 40%
Tabelle 1: Bestandteile von cytoplasmatischen Ribosomen aus Rattenleber. (Quelle: Voet/Voet: "Bio-
chemistry", 2nd ed., John Wiley&Sons Inc.)
Arbeiten mit RNA
Im Gegensatz zu DNA lässt sich RNA basisch hydrolysieren (wegen der 2'-OH-Gruppe). Bei Arbeiten mit RNA ist besondere Vorsicht angebracht. Überall finden sich RNasen (Schweiß, Speichel.), die nur darauf warten, alle RNA-Moleküle zu zersetzen und die
Versuch Nummer FF---009
Arbeit zunichte zu machen. Deshalb müssen während der Arbeiten Handschuhe getra-gen werden. Die Geräte, die man benutzt, müssen auch RNase-frei gemacht werden. Dies erfolgt durch Ausbacken bei 180° C, das Wasser wird mit DEPC (Diethylpyrocar-bonat) und den anderen Lösungen ausgekocht. Dadurch werden die Enzyme, speziell die RNasen, denaturiert (ähnlich der Behandlung mit H2O2). Pipettenspitzen (für Ep-pendorfpipetten) müssen aus frisch geöffneten Tüten genommen werden, oder aus sol-chen, die nur eine kleine Öffnung besitzen. Es werden möglichst wenige Messgeräte be-nutzt; wenn möglich, werden Lösungen zusammen geschüttet und dabei eine Ungenau-igkeit in Kauf genommen. Dabei ist der Kontakt mit den Gefäßrändern zu meiden.
II. Durchführung
Herstellung des Zuckergradienten
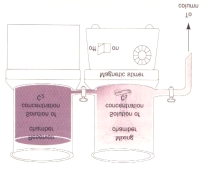
Die Zusammensetzungen der Lösungen sind auf Sei-te 8 zu finden. Für die Trennung der Ribosomenunte-reinheiten benötigen wir einen Gradienten, dessen Dichte zum Röhrchenboden zunimmt. Dieses errei-chen wir, indem wir zwei verschieden konzentrierte Sucrose-Lösungen (30% und 10%) kontinuierlich mit-einander vermischen (siehDie Sucrose-Lösungen werden wie folgt hergestellt: Die ausste-hende 60%ige Stammlösung wird 1:1 bzw. 1:5 mit
RSB-Puffer verdünnt. Für jeden Gradienten, den man Abb. 1: Aufbau zur Herstellung des Sucrose-
erhalten will, müssen je 6 ml beider Lösungen in die Gradienten.
Vorratsgefäße gefüllt werden. Im linken Gefäß befin-
det sich die 30%ige Sucrose-Lösung, im rechten die 10%ige. Sinnvoller Weise öffnet man
zuerst das Ventil, schaltet dann den Rührer ein und danach die Pumpe. Nach einigen
Schwierigkeiten (fehlender Puffer, verstopfte Leitungen ;-) ) hat unsere Assistentin die
Gradienten gegossen. Beim Entfernen der Kanüle muss vorsichtig am Rand entlang ge-
zogen werden, um den Gradienten nicht zu zerstören.
Abtrennung und Aufschließen der Zellen
Aus der HeLa-Stammlösung werden zweimal 50 ml entnommen und bei 25° C für 5 min bei 1000 rpm zentrifugiert. Der Überstand wird abdekantiert (getrennt entsorgen), das Sediment in ca. 1 ml PBS resuspendiert und auf 10 ml aufgefüllt. Nach erneuter Zentri-fugation (25° C, 5 min, 1000 rpm) wird abdekantiert und die Zellenpellets auf Eis gela-gert. Drei Minuten nachdem man die Zellen in 1 ml RSB-Puffer resuspendiert hat, gibt man 2.5 µl 200mM VRC-Lösung hinzu, sowie 55 µl 10% NP40. VRP (Vanadyl-Ribonukleosid-Komplex) inaktiviert Enzyme, v.a. die RNasen; NP40 ist ein Detergens, das die Memb-ran der Tumorzellen auflöst. Man vortext für 30 sec und stellt die Gefäße für dieselbe Zeit auf Eis. Die Zellkerne werden entfernt, indem für 3 min bei 14.000 rpm in der Ep-pendorfzentrifuge zentrifugiert wird.
Isolierung und Trennung der Ribosomen
Eine Grupe trägt 900 µl des Überstandes auf den Gradienten auf. Die andere Gruppe überführt in den Überstand in ein Eppendorfgefäß und fügt 150 µl EDTA zu und gibt davon 900 µl auf den Gradienten. Die anschließende Ultrazentrifugation wird über 3 Stunden bei 35.000 rpm und 4° C mit einem SW41-Rotor durchgeführt. Während dieser Zeit werden die Zellen der HeLa-Stammlösung gezählt. Dazu bringt man 10 µl der Lösung auf eine Zählkammer und wertet unter dem Mikroskop aus.
Versuch Nummer FF---009
Isolierung der ribosomalen RNA
Nach der Zentrifugation wird der Gradient ausgepumpt und die Extinktionen bei
260 nm gemessen; in diesem Bereich absorbieren die aromatischen Ringe der Nukleotide
der (ribosomalen) RNA. Die Messung erfolgt mit Hilfe einer Durchflussküvette, die au-
tomatisch die gemessenen Extinktionen gegen die Zeit (wir haben 10 s eingestellt) aus-
druckt. Etwa 15 Tropfen stellen eine Fraktion dar. Um genauere Aussagen treffen zu
können, wird von den letzten Fraktionen eine Verdünnungsreihe hergestellt. Die Frakti-
onen mit den höchsten Extinktionen werden extrahiert.
Die Extraktion wird mit Phenol und Chloroform durchgeführt. Dabei ist besonders auf
ausreichende Schutzmaßnahmen (Handschuhe, Schutzbrille, Abzug!) zu achten! Phe-
nol und Chloroform sind Gefahrstoffe und bilden ein ätzendes Gemisch! Reste sind
in gesonderten Behältern zu entsorgen! Zunächst aber werden die Fraktionen mit 5 µl
10% SDS und 80 µl 0.2 M EDTA-Lösung versetzt. Die eigentliche Extraktion beginnt mit
der Zugabe von 300 µl wassergesättigtem Phenol und anschließendem Vortexen (Dena-
turierung der Proteine). Es werden 300 µl Chloroform zugefügt (bessere Lösung der Pro-
teine und des Phenols in der organischen Phase) und erneut gevortext. Die Phasentren-
nung erfolgt durch Zentrifugation (2 min, 14.000 rpm, 4° C) in der Eppendorfzentrifuge.
Ein weiterer Extraktionsschritt wird mit der abgenommenen wässrigen Phase durchge-
führt. Nach der zweiten Extraktion werden der wässrigen Phase 20 µl 5 M NaCl zuge-
setzt und gut vermischt. Die Fällung erfolgt über Nacht bei –20° C nach Zugabe von
900 µl absolutem Ethanol. Am nächsten Tag muss die RNA abzentrifugiert (10 min bei
14.000 rpm), mit je 200 µl 70%igem Ethanol gewaschen und erneut zentrifugiert (5 min,
14.000 rpm) werden. Das Ethanol wird abdekantiert und das Pellet in der SpeedVac ge-
trocknet (ca. 30 min).
Für das Gel löst man 0.6 g Agarose in 36 ml H2O und stellt die Mischung für 2 min in die
Mikrowelle (dadurch löst sich die Agarose). 5 ml 10x MOPS und 9 ml Formaldehyd
werden der Lösung zugefügt, wenn diese etwa handwarm ist. Formaldehyd ist giftig!
Nur unter dem Abzug arbeiten! Zuerst werden die Ränder der Gelkammer mit einigen
Tropfen Gellösung abgedichtet, dann der Rest gleichmäßig verteilt und der Kamm ein-
gesetzt. Der Laufpuffer besteht aus 25 ml 10fach MOPS und einem Schluck Formalde-
hyd, die auf 250 ml mit H2O aufgefüllt werden. Im Probenpuffer ist Ethidiumbromid
enthalten, das kanzerogen ist! Es darf nur in Anwesenheit des Assistenten benutzt
werden! Reste sind in gesonderten Behältern zu entsorgen! Um Verunreinigungen zu
entfernen, lässt man das Gel bei 25 V für 30 min vorlaufen.
Die Fraktionen eines Peaks werden in insgesamt 10 µl H2O resuspendiert (erst ein Pellet
resuspendieren, dann mit dieser Suspension das nächste usw.) und mit 2.5 µl 5x Pro-
benpuffer für 10 min auf 70° C erhitzt (Schmelzen der intrachenär zusammengelagerten
RNA). Anschließend werden die Proben wenige Sekunden auf Eis gestellt, um eine er-
neute Doppelstrangbildung zu verhindern.
Die insgesamt 12.5 µl werden komplett in die Taschen eingetragen und dann das Gel ei-
ne Stunde bei 50 V gefahren. Die Auswertung erfolgt unter der UV-Lampe.
Ergebnisse, Auswertung und Diskussion
Die Zellen wurden in zwei Kammern gezählt, die jeweils 0.1 mm tief waren und eine
Fläche von 0.25 mm² aufwiesen. Es wurde nur je ein Viertel der Kammer betrachte; in
der oberen wurden dreimal 18 Zellen gezählt, in der unteren dreimal 23 Zellen. Der
Versuch Nummer FF---009
Mittelwert ist somit
= 4× 20.5 = 82 Zellen. Auf das Volumen bezogen
ergibt sich eine Zelldichte von
= 3.28× 10 Zellen⋅ml-1. Der Wert
0.1 mm × 0.25 mm
stimmt gut mit dem Erfahrungswert von
3.2 × 10 Zellen pro ml überein.
Ultrazentrifugation
Die Beschleunigung (g-Wert) in der Ultrazentrifuge mit dem SW41-Rotor lässt sich ent-weder mit dem im Skript angegebenen Nomogramm oder über Gleichung 1 (S. 1) ermit-teln. Der Außenradius ist r = 15 cm , der Innenradius r = 9 cm . Damit erhält man
• aus dem Nomogramm: 210.000g bzw. 130.000 g
205.400 g bzw. 123.300 g
Extinktionsmessung
Nach anfänglichen Schwierigkeiten ist die Gradientenherstellung und die Ultrazentrifu-gation dennoch gelungen. Im Folgenden sind die Elutionsprofile abgebildet.
Elutionsprofil mit EDTA
Bei diesem Elutionsprofil wird erwartet, dass der erste Peak die undissoziierten 80S-Ribosomen enthält, der zweite die größeren 60S Untereinheiten und im dritten die klei-neren 40S Untereinheiten der Ribosomen vorkommen. Die Auswertung der Gelelek-trophorese (s.u.) sollte daher im ersten Falle sowohl die 28S-als auch die 18S-RNA zei-gen, im zweiten nur die 28S-RNA und im dritten nur die 18S-RNA. Der große Peak zum Ende der Messung hin enthält wahrscheinlich keine native RNA mehr, sondern Lipide, RNA-Fragmente und Proteine.
Versuch Nummer FF---009
Elutionsprofil ohne EDTA
Hier sind zwar drei schwache Erhebungen im Profil zu erkennen, aber sie sind zu schwach, um genügend nachweisbare RNA zu enthalten (vgl. auch Ergebnisse der Gel-elektrophorese). Die letzten beiden hohen Peaks sind gewöhnlich nur, wie im oberen Profil, Nebenprodukte. Dennoch weisen sie die erwarteten 28S- und 18S-RNAs auf. Ein Grund für die späte Elution, d.h. schlechte Zentrifugationstrennung, liegt wahrschein-lich an einer Zerstörung des Gradienten während des Transports oder beim Umgang mit dem Röhrchen. Da die Auswertung über die Zeit und nicht die Fraktionszahl erfolgt, muss aus der Zeit und Gesamtfraktionszahl F ein Zusammenhang zwischen Durchlaufzeit und Fraktion hergestellt werden. Dies geschieht, indem die Zeit t*10s mit dem Verhältnis von Gesamt-fraktionszahl N zu Gesamtzeit T multipliziert wird:
F = t ⋅
Gelelektrophorese
Aufgrund des Elutionsprofils wurden folgende Fraktionen zu Extraktion weiter ver-wendet:
EDTA 12-16 18-22 25-28 29
Die übrigen Taschen wurden nicht beschickt. Dass in Bahn drei dennoch Spuren zu er-kennen sind, liegt an einem Auftragungsfehler; etwas ist von Tasche zwei ausgelaufen. Aufgrund der unterschiedlichen Bandenstärke wurden zwei Fotos mit verschiedener Helligkeit aufgenommen (.
Versuch Nummer FF---009
Abb. 2: Gelfotographie mit starker Helligkeit, um auch die schwachen Banden der Taschen 10
und 12 zu erkennen.
Abb. 3: Gelfotographie mit niedriger Helligkeit, um die Banden der Taschen 2 und 4 besser her-
vor zuheben.
Versuch Nummer FF---009
In der ersten Abbildung erkennt man, dass in nahezu allen Fraktionen beide – sowohl die 18S- als auch die 28S-RNA – enthalten sind. Ausnahmen bilden die Fraktionen der Taschen 8 und 10. In ihnen ist bis auf RNA-Bruchstücke keine auswertbare Ribonuklein-säure nachweisbar. Dies ist nicht unbedingt unerwartet, denn die letzte Fraktion des Gradienten mit EDTA sollte keine RNA enthalten, ebenso erschienen die hohen Peaks des Gradienten ohne EDTA nicht repräsentativ zu sein. Auch hier liegt die Begründung in dem wahrscheinlich verwirbelten Gradienten. Zudem kann die Menge an EDTA noch immer zu gering sein, obwohl sie schon verzehnfacht wurde. Trotzdem ist das Gel gut geworden, denn vor allem die Banden in Tasche 12 sind klar sichtbar. Eine Verbesserung gegenüber früheren Gruppen liegt wohl in dem im Puffer verwendeten, zusätzlichen Formaldehyd und der höheren Menge EDTA begründet.
Zusammensetzung der verwendeten Lösungen
Lösung Konzentration
1.37 M NaCl, 27 mM KCl, 65 mM Na2HPO4, 15 mM KH2PO4
10 mM Tris-HCl pH 7.4, 1.5 mM MgCl2, 10 mM NaCl
0.25 M MOPS pH 7 (Morpholinopropansulfon-
säure), 0.025 M NaOAc, 0.01 M EDTA
1 x Mops, 50% Formamid, 1.8 M Formaldehyd, 3.5% Ficoll 70, 0.025% Bromphenolblau, 0.5 µg/µl Ethidiumbromid
Es wurden folgende Werke als Quellen benutzt: • Voet/Voet: "Biochemistry", 2nd ed., John Wiley&Sons Inc.
• Stryer, "Biochemie", 4. Auflage, Spektrum-Verlag
• CD-Römpp Chemie-Lexikon, Version 1
• Praktikumsvorschrift
Source: http://www.funnycreature.de/RUB/Dateien/BC/BC-FP09.pdf
CHRONIC FATIGUE SYNDROME (Myalgic Encephalitis/Chronic Fatigue Syndrome) ©2012 Keith Berndtson, MD CHRONIC FATIGUE SYNDROME Keith Berndtson, MD Chronic Fatigue Syndrome (CFS) is a difficult-to-treat medical condition whose cause is not yet known. In its 23 October 2009 issue, Science published a study by researchers from the Whittemore-Peterson Institute (WPI), The Cleveland Clinic, and the National Cancer Institute implicating a potential causative role for a retrovirus known as XMRV.1 This study reported that, compared to healthy people, persons with CFS are 18 times more likely to have XMRV DNA in their blood. Retroviruses, including the human immunodeficiency virus (HIV) are know to activate other latent viruses including several from the Herpesvirus family. Unfortunately for sufferers, two years later the XMRV hypothesis went down in flames as several subsequent studies failed to confirm the genetic presence of XMRV in any CFS patients.2 One study claimed to have proved that the original research had found contaminated DNA. Until the fall of 2011, XMRV presented a big challenge to retroviral researchers, as the studies were hard reconcile, meaning one or more of the research teams were getting it wrong. CFS research is famous for producing false leads and misinterpretations of data.3 Nothing gets attention like controversy. CFS is now drawing the attention of a new crop of disciplined, scientific minds. For patients with CFS, and the clinicians who treat them, this is a step in the right direction. On August 23, 2010 a study appeared in the Proceedings of the National Academy of Sciences by Lo and colleagues reporting the presence of DNA fragments from a class of retrovirus known as MLV (mouse leukemia virus). MLVs were present in 85% of 37 CFS patients tested, and in 7% of 44 controls.4 These findings appeared to confirm some role for retrovirus in the pathobiology of CFS, but it was still unknown what role XMRV or MLV retroviruses play in CFS. Some observers felt that these retroviruses interact with other infectious or environmental agents to complicate and worsen CFS severity.5 Some observers were convinced that these viruses play a major causative role in CFS. Still others argued that the data were unclear that these viruses were even present in patients with CFS. In its 1 July 2011 issue, Science published an editorial expression of concern about the October 2009 study.6 In that issue, a study by Tobias Paprotka and a national team of virology experts established that the XMRV DNA found by the WPI team was a
A SHARED DECISION-MAKING™ PROGRAM this program content, including this booklet is copyright protected by health Dialog Services Corporation (HDSC), a related entity of Bupa health Dialog Pty Limited (Bupa Health Dialog), who is licensed to use the material in Australia. You may not copy, distribute, broadcast, transmit, perform or display this program or any part thereof, without permission from Bupa health Dialog. You may not modify the contents of this program without permission from Bupa health Dialog. You may not remove or deface any labels or notices affixed to the program package. © Bupa health Dialog Pty Limited 2012