Case-ha_artikel.pdf
Allergy 2007: 62: 842–856
! 2007 The Authors
Journal compilation ! 2007 Blackwell Munksgaard
Nonallergic angioedema: role of bradykinin
Angioedema is an underestimated clinical problem. Many cases are nonallergic
M. Bas1, V. Adams2, T. Suvorava3,
reactions, e.g. bradykinin-induced angioedema caused by genetic defects and
T. Niehues4, T. K. Hoffmann1,
angiotensin-converting enzyme (ACE) inhibitors. This difference is crucial for
successful therapy, in particular when complete emergency care is not avail-
1Hals-, Nasen- und Ohrenklinik, Universit!tsklinikum
able. Five important forms of nonallergic angioedema can be distinguished:
D"sseldorf, D"sseldorf; 2Herzzentrum Leipzig GmbH,
hereditary (HAE), acquired (AAE), renin-angiotensin-aldosterone system
Universit!tsklinikum Leipzig, Leipzig; 3Institut f"r
(RAAS)-blocker-induced (RAE), pseudoallergic angioedema (PAE) and idio-
Pharmakologie undKlinische Pharmakologie,
pathic angioedema (IAE). Some angioedema are present in the larynx and may
Universit!tsklinikum D"sseldorf, D"sseldorf; 4Klinikf"r Kinder-Onkologie, -H!matologie und -
cause death. A vast majority of nonallergic angioedema are RAE, particularly
Immunologie, Universit!tsklinikum D"sseldorf,
those caused by ACE inhibitors. It appears important to emphasize that in
D"sseldorf, Germany
patients with complete intolerance to RAAS-blockers, cessation of RAAS-blockers is likely to be associated with increased cardiovascular risk. Currently,there is no published algorithm for diagnosis and treatment. Angioedema is
Key words: allergy; angioedema; angiotenin II receptor
usually treated by a conservative clinical approach using artificial ventilation,
type 1 antagonists; angiotensin-converting enzyme
glucocorticoids and antihistamines. Today, a plasma pool C1-esterase inhibitor
inhibitors; bradykinin; C1-INH; ecallantide; icatibant.
(C1-INH) concentrate is the therapy of choice in HAE. The current phar-
Georg Kojda PharmD, PhD
macotherapy of nonallergic angioedema is not satisfactory, thus requiring the
Institut f"r Pharmakologie und Klinische
identification of effective agents in clinical trials. Recently, several new drugs
were developed: a recombinant C1-INH, a kallikrein inhibitor (ecallantide) and
a specific bradykinin-B2-receptor antagonist (icatibant). According to currently
available reports, these drugs may improve the treatment of kinin-induced
Accepted for publication 14 April 2007
Angioedema is a swelling of the mucosa and/or submu-
IAE). A vast majority of these forms of angioedema are
cosa of the skin which may impair breathing and is
induced by increased bradykinin levels (2) and are the
potentially life-threatening (1). Swelling of the head-and-
focus of this review. In contrast, many angioedema must
neck regions, particularly in the pharynx and the larynx,
be classified as IAE. For example, patients with chronic
often require emergency treatment and several days of
urticaria develop nonallergic angioedema in the absence
hospitalization. Other manifestation locations are the
of drug therapy or C1-INH deficiency (3). Pseudoallergic
gastrointestinal tract, the gential region or the extremities.
angioedema is a different form of drug-induced nonaller-
Beside the well-known forms of allergic angioedema,
gic angioedema and is mediated by a so-called pseudo-
many forms of nonallergic angioedema are known and a
allergic process which is presumably linked to the
vast majority of these are caused by increased plasma and
mechanism of action of the triggering drug. For example,
tissue concentrations of bradykinin. Thus, these forms of
the pseudoallergic reaction to aspirin is thought to be a
angioedema share a similar pathophysiology.
result of the inhibition of cyclo-oxygenase and subse-quently increased generation of cysteinyl-leukotrienes (4).
Forms of angioedema
Epidemiology of angioedema
In general, angioedema can be allergic or nonallergic,which basically means immunoglobulin (Ig) E-mediated
In people with HAE, heterozygous C1-INH deficiency
or not IgE-mediated, respectively. Nonallergic angioede-
results in autosomal-dominant inheritance with an inci-
ma might be caused by hereditary disposition or is of
dence of 1:50 000 and there are no differences depending
iatrogenic origin, and can be divided into five different
on ethnic groups or sex (5). In contrast, AAE is a rare
types (Fig. 1): hereditary (HAE), acquired (AAE),
condition (6, 7). The incidence of RAE induced by ACEi
RAAS-blocker-induced (RAE), pseudoallergic angioede-
has been estimated with great variety which is most likely
ma (PAE) and idiopathic angioedema (unknown cause,
due to race differences. For example, in white Caucasians,
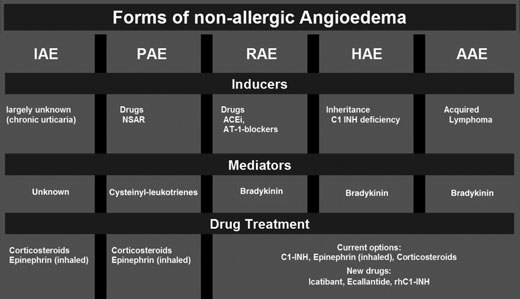
Nonallergic angioedema
Figure 1. Different forms of angioedema (HAE: hereditaryangioedema, PAE: pseudoallergic angioedema, RAE: RAAS-blocker-induced angioedema, AAE: acquired angioedema).
the frequency of ACEi-induced angioedema is reported to
mouse chromosome 7 (16). It is expressed in several
range between 0.1% and 0.7% (1, 2, 5, 8, 9), while Black
tissues like kidney, blood vessels, pancreas, gut salivary,
people show a much greater susceptibility (10). In fact, a
spleen, adrenal and neutrophils (17–20). From a cardio-
recent meta-analysis investigating adverse reactions to
vascular viewpoint, the kallikrein-kinin system is thought
drugs used in cardiovascular medicine found a relative
to antagonize the effects of the RAAS, and is closely
risk of 3.0 for the development of ACEi-induced angi-
related to this system (Fig. 2). The functional coupling
oedema among Black compared to White people (11).
between the two systems is illustrated by angiotensin-
Based on roughly 6.5 million users of ACEi in
converting enzyme (ACE) whose two active sites are able
Germany and on an average frequency of angioedema
to generate angiotensin II from angiotensin I and to
of 0.3%, approximately 20 000 cases of ACEi-induced
degrade kinins into inactive peptides (21, 22).
angioedema will be expected to occur. Thus, the calcu-lated incidence approaches 1 : 4000 demonstrating that
Bradykinin receptors and signal transduction
ACEi-induced angioedema, which represents the majorityof RAEs, appears to be much more frequent than HAE
Kinin receptors are cell surface, G-protein-coupled re-
(12). Interestingly, AT-1-blockers appear to induce RAE
ceptors of the seven-transmembrane family. So far, two
but with a lower frequency than ACEi (2) and this
subtypes of the receptor – the bradykinin receptor type 1
estimation has been confirmed in large clinical trials (13).
(BKR-1) and the BKR type 2 (BKR-2) – are identified,
There are no data on the epidemiology of angioedema
based on their pharmacological properties (23–26) and on
caused by pseudoallergic reactions to drugs.
expression cloning (27–29). The human BKR-2 gene islocated on chromosome 14q32 (30), whereas the BKR-1has been mapped to chromosome 14q32.1–q32.2 (31). At
Bradykinin in the human body
the amino acid level, the BKR-1 and the BKR-2 shareonly 36% sequence homology (29). The BKR-1 is
The kallikrein-kinin system
synthesized in a variety of different organs de novo
The discovery of the kallikrein-kinin system dates back to
following tissue injury (31, 32), whereas the BKR-2 is
1909, when Abelous and Bardier demonstrated the
constitutively expressed in a larger number of tissues (33).
hypotensive effect of urine (14). Kinins are pharmaco-
Bradykinin is thought to be one of the most potent
logically active peptides released into body fluids and into
vasodilatators, as it is capable to liberate three important
tissues as a result of the enzymatic action of kallikreins on
endothelium-derived vasodilatory mediators: NO, pro-
kininogens. Kinins are a family of peptides, including
stacyclin (PGI2) and endothelium-derived hyperpolariz-
bradykinin, kallidin and methionyl-lysyl-bradykinin, of
ing factor (EDHF; 34). It is generally accepted that the
which kallidin and methionyl-lysyl-bradykinin are con-
activation of the BKR-2 on endothelial cells leads to an
verted very rapidly into bradykinin via the action of
activation of phospholipase C gamma via a transient
aminopeptidases present in the plasma and urine (15).
Tissue kallikrein (EC 3.4.21.35), which differs signifi-
increased formation of inositol 1,4,5-triphosphate (IP3)
cantly from plasma kallikrein, is encoded by the KLK1
and diacylglycerol (35, 36). As a consequence of an
gene located on human chromosome 19q13.2–q13.4 and
elevated IP3 concentration, intracellular calcium rises by
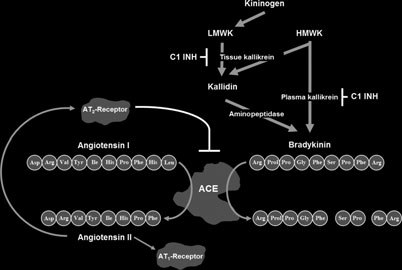
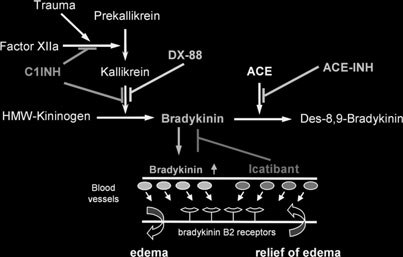
Scheme of the connection between the renin-angio-
Detrimental and beneficial effects of activation of
tensin-aldosterone system and the kallikrein-kinin system.
BKR-2 in humans.
activates protein kinase A leading to an acute increase ofNO due to a phosphorylation of eNOS at Ser1179 (40).
Physiological actions of bradykinin
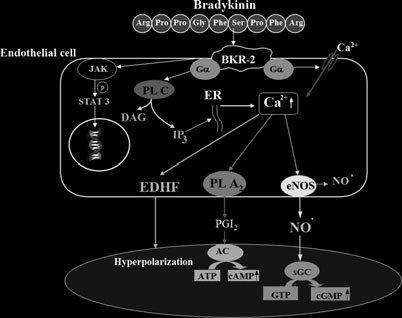
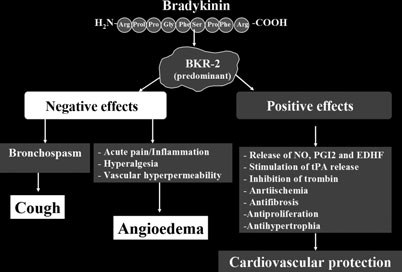
Much progress in understanding the physiological roleof kinins has been made in the 1980s, when differ-ent selective antagonists of BKR-1 and BKR-2 werediscovered (26). More recently, the development ofC1-INH- and BKR-2-transgenic mice provided import-ant knowledge concerning the role of kinins in vivo (41,42). The effect of bradykinin on vascular permeabilitywas demonstrated by targeted disruption of C1-INH(42). Bradykinin was shown to dilate peripheral andcoronary vessels, may decrease arterial blood pressurein normotensive animals, and exert antithrombogenic,antiproliferative and antifibrogenic effects (Fig. 4; 33,43–47).
Scheme of the mechanisms of action of bradykinin-
Cardiovascular actions of bradykinin are believed to be
induced activation of vascular endothelial cells. Mediators such
mainly mediated by the activation of BKR-2 on endot-
as nitric oxide and prostacyclin act in an autocrine and para-
helial cells leading to the release of NO, PGI2 and EDHF
crine manner.
(48–52) and to the liberation of tissue plasminogenactivator (53). Bradykinin is also shown to be involved
liberalization from internal stores or by an increased
in the cardioprotective effect of preconditioning on
Ca2+ influx (37), finally leading to an activation of the
myocardial ischaemia/reperfusion injury (54). It can
Ca2+-sensitive endothelial nitric oxide synthase (eNOS).
reduce the infarct area (55, 56) and has a growth
In addition, the elevated intracellular Ca2+ activates the
inhibitory effect to cardiomyocytes (57, 58). Additionally,
Ca2+-sensitive phospholipase A2, which hydrolyses mem-
kinins can evoke contractions of human bronchi smooth
brane phospholipids liberating arachidonic acid, which is
muscles (59), suggesting that bradykinin mediates dry
the rate-limiting step in the synthesis of PGI2 (38).
cough induced by ACEi. Indeed, experimental data
Beside these calcium-regulated signalling pathways,
suggest that bradykinin might be a key substance in the
recent studies also explored the importance of signalling
pathomechanism of coughs associated with ACE inhib-
pathways depending on phosphorylation. Ju and col-
itors (60, 61). In addition, local accumulation of bradyk-
leagues demonstrated that bradykinin activates tyrosine
inin may lead to the activation of proinflammatory
kinase 2 (Tyk2) of the Janus-activated kinase (JAK)
peptides and local release of histamine, inducing a cough
family, resulting in subsequent tyrosine phosphorylation
reflex hypersensivity (62).
and nuclear translocation of STAT3 (39). Using similar
The kallikrein-kinin system also plays an important
approaches, it could be demonstrated that bradykinin also
role in handling sodium and water metabolism. The
Nonallergic angioedema
natriuretic and diuretic effects of endogenous kinins are
underlying mechanisms were extensively discussed and
documented by several approaches, including BKR-2
reviewed previously (9, 83). Briefly, disrupted inhibition
gene inactivation in transgenic mice: these animals are
by C1-INH deficiency of the serine proteases C1s and C1r
prone to salt-sensitive hypertension (63, 64). However,
promotes activation of the complement system. In addi-
this effect of bradykinin could not be confirmed in
tion, the inhibition by C1-INH of two proteases of the
another knockout strain (64). There is compelling evi-
coagulation system, namely factor XIIa (Hagemann
dence linking bradykinin to pathophysiological processes
factor) and kallikrein appears important. As discussed
that induce tissue damage and inflammation, hyperaemia,
above, the kallikrein activity is a major player in the
leakage of plasma proteins, bone resorption induced by
synthesis of bradykinin. In particular, the vasodilator
inflammation and pain; besides, many of these activities
activities of bradykinin and its potency to promote and
follow stimulation of BKR-1 (65–71). Bradykinin is also
induce increased vascular permeability are well consistent
involved in the production of pain and hyperalgesia by
with its putative role in the pathophysiology of angi-
direct activation of BKR-2 receptors on primary nonmy-
elinated sensory neurones and thus participates in the
According to our current knowledge, a deficiency of
direct pain response (65). When inflammation is pro-
C1-INH due to genetic defects plays a causal role in HAE
longed, BKR-1, which are not expressed in healthy tissues
(9). The human C1-INH gene has been mapped to
to a significant degree, also play an important role in the
chromosome 11 (11q12–q13.1; 5). Two variants of HAE
maintenance of hyperalgesia. In vivo evidence for this was
have been described: HAE type 1 with decreased C1-INH
obtained in BKR-1 knockout mice which exhibited a
levels and functional deficiency (85% of cases) and HAE
reduced inflammatory response and hypoalgesia (72, 73).
type 2 with normal protein concentration but a functional
Transgenic mice with overexpression of BKR-1 devel-
defect (15% of cases; 84). Recently, a first case of
oped exacerbated paw oedema, induced by polysaccha-
homozygous C1-INH deficiency with the mutation c.
1576T>G was reported (85). Targeted disruption of C1-
lipopolysaccharide-induced endotoxic shock, supporting
INH in mice resulted in increased vascular permeability
the notion that BKR-1 plays an important role in
as evidenced i.v. injection of Evans blue in the tail and
modulating inflammatory responses (74).
this was reversed by treatment with human C1-INH (42).
Finally, bradykinin has been shown to increase the
Other data obtained with this transgenic mouse strain
release of insulin from pancreatic b-cells through the
provided strong additional evidence for a crucial involve-
increase of intracellular calcium in response to hypergly-
ment of BKR-2 in the pathogenesis of angioedema. For
caemia (75, 76) and enhance insulin-dependent glucose
example, increased vascular permeability was strongly
transport (77). Other data revealed that bradykinin
reduced by treatment with the BKR-2 antagonist icatib-
released locally can regulate the uptake and availability
ant or by simultaneous homozygous disruption of the
of glucose in target tissues independently of the release of
insulin (78, 79). These data suggest that decreaseddegradation of bradykinin contribute to the beneficial
Acquired angioedema
effects of ACEi in cardiovascular patients, such asreduced complications related to diabetes and a reduction
AAE develops on the basis of nongenetic C1-INH
of new cases of diabetes type 2 (80).
deficiency, which primarily affects adults or elderlypatients (9). This might be a result of various conditions,including severe illness such as malignancies. Patients
Pathophysiology of bradykinin-induced angioedema
with lymphoproliferative diseases may develop angioede-ma associated with decreased C1-INH plasma levels and
Hereditary angioedema
activity (7, 86). In contrast to HAE where the synthesis of
In 1882, Heinrich Irena¨us Quincke described an acute and
C1-INH is defective, AAE is characterized by large
clearly circumscribed oedema. Although other case
reports on such oedema had already been published, it
(autoantibodies) consuming the C1q molecules and sub-
was his real reward having accurately described this
sequently C1-INH (87). Thus, C1q levels are normal in
disease and separated it from urticaria (81). Today, the
HAE, but decreased in AAE. In addition, C4 levels are
so-called Quincke oedema is synonymous with angioede-
low and C3 levels are normal in AAE. Other diseases such
ma, and is still used to describe a circumscribed oedema
as hepatocellular carcinoma and liver cirrhosis might be
without urticaria and/or pruritus.
associated with decreased C1-INH plasma concentration
It is known that the serine protease inhibitor (serpin)
or activity, but subsequent angioedema has not been
C1-INH has a variety of biological activities (82). Of
described (88, 89). There is also a report on a lymphoma-
these, the disrupted inhibition by C1-INH deficiency of
associated angioedema with a normal plasma concentra-
several proteases of the complement and the contact
tion of C1-INH (90), and a recent characterization of a
system and of kallikrein appear to be of particular
new C1-INH mutation, leading to strong inhibition of
importance for the development of angioedema. The
monocyte C1-INH secretion (91).
treatment in severely ill cardiovascular patients who
developed RAE. However, new s.c. drugs emerging to
An essential component of the RAAS is ACE (carboxyp-
treat angioedema (see below) are being evaluated in HAE
eptidase, kininase 2, EC 3.4.15.1), an enzyme with two
but not in RAE.
major proteolytic tasks: generation of angiotensin II anddegradation of bradykinin. In addition, ACE also
Factors triggering angioedema attacks
degrades substance P (92, 93). Treatment with ACEiincreases the plasma levels of bradykinin and the biolo-
Numerous factors have been reported by patients with
gical activity of bradykinin is crucially involved in the
HAE as triggers or inducers of angioedema attacks.
actions of ACEi. For example, it has been shown that
These include exposure to cold, mechanical trauma, tissue
permanent blockade of BKR-2 by icatibant (formerly
compression, prolonged sitting or standing, certain foods
Hoe 140) halved the reduction of blood pressure induce-
(e.g. eggs), infections, concomitant diseases, contact to
able with captopril in hypertensive patients (94). ACEi-
pesticides or chemicals either directly or via new products
induced angioedema are perhaps caused by decreased
or clothes, excitement/stress and certain drugs, such as
degradation of bradykinin (95). Interestingly, combined
ACEi and oestrogens (9). However, these anecdotic
inhibitors of neutral endopeptidase and ACE, such as
reports have never been systematically investigated and
omapatrilat showed a dramatic fourfold increase of
appear to depend on the patient!s individual character-
angioedema when compared to the ACEi enalapril
istics. One exception is the use of contraceptive oestro-
(2.17% vs 0.68%; 96). Bradykinin might also be involved
gens and hormone replacement therapy with oestrogens.
in dry cough, a typical side effect of ACE inhibitors (93).
It has been shown that some female HAE patients
AT-1-blockers appear to induce angioedema with a
respond to physiologically (first manifestation after
lower frequency than ACEi (2, 13). Again, these angi-
menarche, menstrual cycle and pregnancy) and/or
oedema appear to be caused by bradykinin. It was shown
pharmacologically increased serum oestrogen (contra-
recently that AT-1-blockers increase bradykinin levels in
ceptive oestrogen/gestagen combinations and hormone
hypertensive patients (97). This effect was associated with
replacement therapy with oestrogen) with an increased
an increased ratio of bradykinin and its degradation
frequency of attacks (104, 105). Other researchers found
product BK1-7, suggesting that AT-1-blockers may
similar cases in men and women after antiandrogenic
inhibit ACE by a mechanism different from ACE-
treatment with cyproterone (106). Likewise, a significant
inhibitors. The circulating levels of angiotensin II increase
correlation between the attack frequency and serum
in response to AT-1-blocker therapy, because these drugs
progesterone levels was evident in 44 female HAE
interrupt the physiological feedback mechanism regula-
patients, but not in the 34 male patients of this study
ting angiotensin II synthesis by the release of renin (98).
cohort (107).
At the same time, all other AT-1 receptors are also
The molecular mechanisms of triggering factors are
blocked, leaving much room for the activation of
largely unknown. One exception is the use of ACEi in
angiotensin II type 2 receptors, which, in turn, might
HAE patients, which further raises plasma and tissue
inhibit the degradation of bradykinin by inhibition of
bradykinin (see above). Thus, it is not surprising that
ACE or of neutral endopeptidase (97). However, previous
HAE patients sensitively respond to ACEi with an
animal studies showed the stimulation of angiotensin II
increase of their attack frequency (108, 109). A similar
type 2 receptors might actually stimulate bradykinin
mechanism is probably driving angioedema attacks in
production so further studies are required to justify the
cardiovascular patients without HAE. However, import-
hypothesis given above (99). Nevertheless, AT-1-blockers
ant questions remain to be clarified. For example, why do
should be used with caution in patients who have
only <1% of the patients taking ACEi develop
experienced ACEi-induced angioedema (100).
angioedema, while bradykinin levels are increased in all
ACEi and AT-1-blockers are widely used as level I
ACEi-patients? Furthermore, what is the reason that
evidence drugs to treat hypertension, myocardial infarc-
some patients develop angioedema within a few days after
tion, heart failure and type I diabetic nephropathy (101–
starting the ACEi therapy, whereas others have their first
103). The beneficial effects of these drugs in heart failure
attack after years of uneventful treatment (110)?
and after a myocardial infarction include improvements
It might be speculated that a variety of endogenous
in survival, the rate of hospitalization, symptoms, cardiac
factors counterbalance angioedema-inducing biological
performance, neurohormonal levels and reverse remod-
activities of bradykinin. In contrast, other factors might
elling (101, 102). Thus, in patients who develop RAE, the
create a tissue environment that disrupts this balance and
cessation of RAAS-blocker therapy is, over the long term,
induces angioedema. As many of the above-mentioned
likely to be more important problem than is angioedema
triggering factors are associated with an activation of
itself. The availability of an easy-to-use oral or s.c. stand-
inflammatory pathways, it appears promising to look for
by medication which can be self-administered whenever
an association between inflammatory plasma markers and
an attack is recognized by the patient might be a
the development of angioedema. Recent findings may
promising future approach to maintain RAAS-blocker
shed some light on such potential trigger factors. In a
Nonallergic angioedema
Figure 5. Plasma levels of (A) C-reactive protein and (B) fibrinogen in patients with RAAS)-blocker-induced caused by ACEi andpatients with angioedema of unknown cause [data adopted from Ref. (111)].
cohort of 43 non-HAE male and female patients suffering
from an acute angioedema attack, 25 were taking ACEiand their attack completely resolved following drug
initial emergency care
cessation, while the other 18 cases showed no physical,biochemical or genetic signs allowing classification of their
Monitoring vital functionsRating angioedema status
attack and were thus assigned to the group of IAE.
Supplying OxygenEmergency pharmacotherapy
Determination of acute-phase proteins during the acute
Intubation, tracheotomy
attack revealed striking differences between these two
groups (Fig. 5). Patients on ACEi treatment had strik-ingly and significantly increased plasma levels ofC-reactive protein and of fibrinogen (111).
if needed laryngoscopy
It should be added that in all these ACEi-treated
patients, the levels of acute-phase proteins returned to
normal as measured 3–6 months after the attack. Fur-thermore, the determination of acute-phase proteins in 21cardiovascular patients treated with ACEi who never
experienced angioedema showed normal values as well.
A subsequent laboratory investigation showed not only
potent in vitro vasodilator effects of fibrinogen in human
artery rings, but also a strong potentiation of the
Figure 6. Algorithm for the diagnosis of angioedema.
vasodilatory efficacy of bradykinin (M. Bas and G.
Kojda, unpublished results). Taken together, these resultsmay point to a contribution of the inflammatory process
patient!s family history and current medications should
to the pathophysiology of angioedema. However, it is still
be performed.
not clear whether or not acute-phase proteins are truly
A detailed physical examination should reveal whether
the swelling is itchy or painful, which indicates allergicangioedema or inflammatory conditions, respectively.
Signs of urticaria usually exclude nonallergic angioede-ma. Blood measurements must include the determination
Basic principles of diagnosis
of C1-INH activity, and, eventually, C1-INH concentra-
One of the most important procedures to diagnose
tion and markers of inflammation (e.g. C-reactive
oedema (see Fig. 6) is to separate allergic from nonaller-
protein, leucocyte count). A reduced activity of plasma
gic angioedema and to exclude other pathologies such as
C1-INH is indicative of either hereditary or AAE, and
infection, inflammation, tumours and diseases of large
should be examined further, e.g. by genotyping or
salivary glands. These procedures usually follow the
determination of accompanying diseases such as lym-
initial emergency care, e.g. monitoring of vital functions,
phoma (please see "Case report: Acquired angioedema!).
determination of the status of angioedema, intubation,
In some assumed drug-induced cases (ACEi), it might
oxygen supply and pharmacotherapy with antihistamines,
also be helpful to determine serum ACE activity. As for
nebulized adrenaline and corticosteroids (1, 9). Beside a
diagnostic imaging procedures, ultrasound visualization
of the oedema region is usually sufficient, but uncertain
laryngoscopy, several blood measurements, some imaging
cases and/or abdominal pain should undergo magnetic
procedures and an extensive anamnesis focusing on the
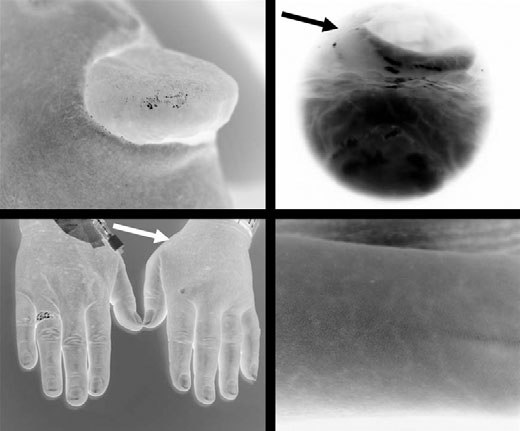
Figure 7. (A) ACEi-induced angioedema of the tongue. (B) Supraglottic laryngeal oedema caused on AT-1 R-blocker. (C) Hereditaryangioedema of the left hand. (D) Typical dermal efflorescence in HAE.
A first suspicion of HAE is given by mucosal
tine 2 mg), i.v. glucocorticoids (e.g. methylprednisolone
swellings and/or unclear abdominal pain in patients at
250–1000 mg) and/or adrenaline inhalation. The appli-
a young age and those with a family history, a lack of
cation of antihistamines in nonallergic angioedema may
concomitant drug therapy and the absence of urticaria
work in IAE but is ineffective and hence superfluous in
as well. Following eventually necessary emergency
RAE, AAE and PAE (2). This therapy is due to the fact
treatment, the diagnostic procedure for HAE (type 1
that in an emergency situation – a patient with poten-
and type 2) requires the measurement of both C1-INH
tially life-threatening dyspnoea – the doctor may not be
serum concentration and function (112). Furthermore,
able to entirely exclude an allergic background. Simi-
plasma levels of the complementary factor C4 in the
larly, glucocorticoids are not proven to be effective in
serum are reduced in a vast majority of the cases. This
kinine-induced angioedema (108). However, glucocortic-
factor is frequently reduced to approximately 25% of
oids still belong to the standard for treatment of
the normal value during the acute episode of angioede-
angioedema (114). Principally, this medication should
ma (81, 113). Furthermore, other rare conditions, such
lead to a decrease in mucosal swelling. However, a
as Crohn!s disease of the mouth and lips, facial
clinical study showing a beneficial effect in patients with
cellulitis and the superior vena cava syndrome should
angioedema is lacking. It also has to be considered that
be excluded (5). An illustration of different manifesta-
this medication is used "off label! and may induce adverse
tions of bradykinin-induced angioedema is depicted in
effects, e.g. increased blood pressure. Thus, consequent
monitoring of blood pressure and pulse are required.
Inhaled adrenaline may also be effective in laryngealoedema or other severe forms, but this is not based on
Current treatment of angioedema
evidence and adrenaline inhalation is not labelled for thisindication.
Basic emergency treatment
In case of a progressive course of pharyngolaryngeal
Depending on the symptoms and clinical findings,
angioedema, it may be necessary to temporarily bypass
emergency treatment consists of oxygen application,
the airflow via intubation or tracheotomy. During the
pharmacotherapy, intubation and, in severe cases, a
acute angioedema episode, it is technically difficult to
tracheotomy (84). Currently, in most medical institu-
insert a ventilation tube and thus this attempt should be
tions, pharmacotherapy of acute pharyngolaryngeal
performed in the presence of an experienced head-and-
angioedema consists of i.v. antihistamines (e.g. clemas-
neck surgeon able to quickly perform a tracheotomy.
Nonallergic angioedema
In cases with extensive cervical swelling it might be
antihypertensive drugs. As described above, AT-1-block-
difficult to identify the trachea and thus an emergency
ers should not be used routinely to replace the much more
coniotomy (between the cricoid and thyroid cartilage)
commonly used ACEi (please see "Case report: RAAS-
might become necessary. Taken together, the current
blocker-induced angioedema!) (100, 119, 120). For exam-
pharmacotherapy of acute nonallergic angioedema is not
ple, the patient described in the RAE case below received
satisfactory, thus requiring the identification of effective
amlodipin to replace candesartan, and this might be
agents in clinical trials.
sufficient, as long-acting dihydropyridines have beneficialeffects on the prognosis of patients with hypertension(103). However, this is strikingly different in patients with
Hereditary and acquired angioedema
a history of myocardial infarction or heart failure (101),
As extensively reviewed recently (84), both types of
where no equally effective drug is available. One possible
angioedema are characterized by C1-INH deficiency, and
approach to treat such patients in the future would be to
usually respond rapidly to treatment with therapeutic
provide a safe and easy-to-use stand-by medication which
C1-INH, which is currently the "gold standard! of
can be self-administered whenever an attack is scented by
pharmacotherapy. It is not available in the USA, where
the patient.
HAE is treated with the alkylated androgens stanazololand danzol (see Case report AAE). This i.v. substitutiontherapy with C1-INH concentrate derived from human
Case report: Acquired angioedema
plasma pools might also be used to prevent acuteangioedema attacks (9). Although there is theoretical
A 65-year-old female patient with a history of surgically
risk of infection with viruses such as HIV, the product has
treated Morbus Crohn presented at the emergency
been proved to be safe, as there is not one known case of
department of the university hospital in Duesseldorf,
viral infection despite many million applications. How-
Germany, with recurrent angioedema located on her right
ever, in view of the uncomfortable i.v. application route
arm, hips and legs. A detailed diagnostic procedure
this approach may be difficult to manage for the patient
revealed a strongly decreased C1-INH activity (27%,
(9). In contrast, drugs such as danazol (see Case report
norm: 70–100) but no mutations in the C1-INH gene.
AAE) and tranexamic acid are available in oral form, and
Other abnormal laboratory findings were: increased C-
are used as a preventive pharmacotherapy to reduce the
reactive protein of 1.3 mg/dl (norm: <0.5), increased
frequency of attacks.
fibrinogen of 471–530 mg/dl (norm: 180–350) and in-
Tranexamic acid is a fibrinolytic drug used for short-
creased alkaline phosphatase of 120 U/l (norm: 35–104).
and long-term prophylactic pharmacotherapy in HAE, as
Further examinations were carried out to look for
it can reduce the number and severity of attacks in HAE
lymphoproliferative diseases which have been reported to
(113). Tranexamic acid is structurally related to the
induce C1-INH deficiency (see above). Computerized
amino acid, lysin and saturates lysin-binding regions in
tomography showed no signs of splenomegaly or abnor-
plasminogen by forming a reversible complex. This
mal lymph nodes, and blood leucocyte counts were
inhibits the binding of plasminogen and plasmin to fibrin,
normal. A bone marrow biopsy revealed an infiltration
disturbs the proteolytic activity of plasmin and subse-
with a non-Hodgkin lymphoma. Histology showed a
quently inhibits fibrinolysis (115). An accompanying
20% CD20- and CD43-positive infiltration and a cell
effect of this action of tranexamic acid is the reduced
population expressing CD23 and CD38. A bone marrow
consumption of C1-INH, and this effect is used in the
B-cell non-Hodgkin lymphoma was diagnosed. A specific
treatment of HAE (9, 113). The potency of tranexamic
treatment was not necessary, but the patient was advised
acid in preventive pharmacotherapy in HAE patients is
to return for further bone marrow biopsy and to avoid
reported to be low, particularly among children (113,
ACEi. Her acute angioedema attacks were treated with
116). Its important side effects include gastrointestinal
C1-INH concentrate and danazol (200 mg/day) was
symptoms, such as nausea, vomiting, diarrhoea and rare
prescribed to prevent further attacks. Her repeatedly
cases of thrombotic/embolic events (117, 118). In addi-
measured C1-INH plasma concentration after 20 months
tion, visual impairment and vertigo have been noted after
of danazol therapy was 0.11 g/l (norm: 0.17–0.44) and
long-term use of tranexamic acid. An annual fundoscopy
C1-INH activity was 34% (norm: 70–100). Despite this
is a recommended check for any impairment of visual
apparent lack of a danazol-effect on C1-INH concentra-
function, including retina tumours (113).
tion and activity, the patient experienced no furtherangioedema attacks. The non-Hodgkin lymphoma didnot progress after that.
RAE is basically treated as described in the section Basic
Emergency treatment (see above). Furthermore, theimmediate cessation of RAAS-blockers is mandatory.
Angioedema might be the first sign of a lymphoprolif-
Later on, the RAAS-blockers need to be replaced by other
erative disease, as was evident in the present case. In this
case, successful treatment with danazol, a steroid which
blood pressure was 130/80 mmHg, and he was still taking
stimulates the synthesis of C1-INH, and of C4 and
phenprocoumon (INR 3,1), sotalol mite (160 mg b.i.d.),
which is capable of reducing the frequency of angioede-
l-thyroxine (150 mg daily) and tamsulosin (0.4 mg daily).
ma attacks in AAE (121), was successful. The drug is
available in several European countries and in the USA
(20 mg/day) was initiated. A routine laboratory investi-
but no longer in Germany. In the USA, danazol and its
gation on admission showed increased values of fibrin-
congeneer, stanozolol, are widely used to treat angi-
ogen (549 mg/dl; norm: 180–350) and leucocytes (11 900/
oedema, including acute attacks in children. Interest-
ll; norm: 4000–11 000/ll). All other routinely determined
ingly, this case also disputes the assumed mechanism of
values were normal.
action of danazol and indicates that an increase of
The patient was again treated with 250 mg i.v. meth-
C1-INH concentration and activity are accompanied by
ylprednisolone, 2 mg i.v. clemastine, 5 mg nebulized
other yet unknown activities which interrupt the path-
adrenaline and oxygen supply and recovered within
ophysiology of angioedema. For example, in women,
24 h. On being discharged, he was advised to avoid both
danazol induces deprivation of oestrogen, a well-known
types of RAAS-blockers, ACEi and AT-1-blockers.
trigger of angioedema. Another interpretation is that
Subsequently, candesartan was replaced by 5 mg/day
danazol is not the underlying cause for the cessation of
amlodipin, 50 mg/day triamterene and 25 mg/day hydro-
angioedema. It is known that angioedema might not
occur for many months despite continuously decreasedplasma C1-INH (9). However, long-term use of danazol
may induce rare but severe side effects, e.g. liver diseasessuch as hepatic necrosis or cholestasis, adenomas and
This case demonstrates that RAE can include severe
eventually carcinomas (9, 122). Of the many other side
laryngeal oedema, which can induce life-threatening
effects, some may result from oestrogen deprivation in
breathing impairment eventually requiring tracheotomy
(110). Furthermore, this case is an example for recurrentRAE when an ACEi was replaced by an AT-1-blocker.
In a large controlled clinical trial the frequency ofangioedema induced by captopril (0.5%) was higher than
Case report: RAAS-blocker-induced angioedema
that induced by a valsartan (0.2%) (13). However,
A 62-year-old male patient presented to the emergency
department with dyspnoea and diffuse swelling of his
might be more susceptible to angioedema induced by
tongue that had developed within a few hours. Transnasal
AT-1-blockers (see section RAAS-blocker-induced angi-
flexible endoscopy revealed an oedema of the hypophar-
oedema), although previous investigations on this matter
ynx. He had no known history of allergies. He was taking
do not support this view (99, 123). Thus, further clinical
enalapril (5 mg daily) for the past 7 years to treat
studies are necessary to estimate this risk more accurately.
hypertension (admission blood pressure 130/80 mmHg).
However, it appears necessary to precisely inform about
Furthermore, the patient was wearing an atrial pacema-
this potentially life-threatening side effect, including a
ker, had an operation with aortic valve replacement
clear information for patients. Finally, this case matches
7 years ago and was suffering from hypothyroidism.
similar cases registered worldwide and support the
Additional prescription drugs are: phenprocoumon (INR
2,7), sotalol mite (160 mg b.i.d.), l-thyroxin (150 mg
caution in patients with previous ACEi-induced angi-
daily), tamsulosin (0.4 mg daily). Laboratory values on
oedema (124).
admission showed a low increase of the acute phaseproteins CRP (0.8 mg/dl; norm: <0.5) and fibrinogen(370 mg/dl; norm: 180–350). All other routinely deter-
Special considerations in paediatric treatment of
mined values were normal.
The patient was treated with 250 mg i.v. methylpredn-
isolone, 2 mg i.v. clemastine, 5 mg nebulized adrenaline
In children, the main cause of kinin-induced angioedema
and oxygen supply. He recovered within 24 h despite his
is HAE. Insufficient C1-INH function as a result of
severe hypopharyngeal oedema. On discharge, he was
autoimmunity is also rare in children. It is likely that
advised to avoid enalapril and other ACEi. Instead,
HAE is under-diagnosed in childhood and adolescence.
therapy with 5 mg amlodipin was initiated to treat his
It is estimated that 85% of the patients with HAE show
symptoms before they are 20 years old. In contrast, only
After 24 months, the patient returned, but with a
35% are diagnosed at that age. The clinical diagnosis of
supraglottic laryngeal angioedema, accompanied by dys-
HAE is difficult in children because they may present with
pnoea and dysphonia. In the meantime, his antihyper-
symptoms that are very common in childhood. Among
tensive medication was changed from amlodipine to
these symptoms is: abdominal pain, swelling of the
12.5 mg/day of AT-1-blocker candesartan. His admission
extremities, difficulties to swallow, hoarseness or acute
Nonallergic angioedema
dyspnoea. Unless there is a positive family history, these
convenient for the use in long-term prophylaxis. Choices
symptoms are not immediately recognized as symptoms
to C1-INH include antifibrinolytic agents like tranexa-
of HAE, and, therefore, there is a diagnostic delay.
mic or !-aminocaproic acid or danazol. The antifibrin-
Evidence for treatment relies mainly on data from a few
olytic agents appear not to be an alternative because
registries that report their paediatric data or consensus
they cause adverse effects like myalgias and myonecrosis,
conferences (9, 84, 113).
abdominal pain, hypotension and thromboses. In astudy by a Hungarian group, 26 children from 1 to15 years of age are reported (9, 116). Of them, 11
Education of parents and the patients (once they are adolescents)
children were put on long-term prophylaxis with 1–2 g
First of all, medical education is of key importance. If
tranexamic acid because of frequent or life-threatening
the parents are informed about the disease early diag-
attacks. This proved to be ineffective in eight of the 11
nosis will be possible and unnecessary operations like
and, therefore, the remaining eight children where put
appendectomy in case of intra-abdominal oedema will be
on danazol 100–200 mg/day, which the authors report to
avoided. This point cannot be stressed enough, because
be effective and without serious side effects (with the
in our own experience, unnecessary operations have been
exception of one girl with a delayed menarche). How-
performed although parents knew about the diagnosis of
ever, this series reports only a small number of children
HAE in their children but not about their possible
(n = 8) and the potential long-term side effects of
manifestations. Avoidance of oral contraceptives in
androgens in children (premature epiphysial closure with
adolescent girls should be a part of counselling.
growth arrest, liver damage and hepatocellular adenoma,
An important part in the education of patients is the
development of secondary sexual characteristics, hirsu-
knowledge of and contact with patient initiatives, in
itism and many more) make many paediatricians very
particular self-regulating communities which can be
cautious about their use.
easily identified by their web pages. Parents need to be
In conclusion, appropriate education of patients and
provided with emergency lots of C1-INH concentrate for
parents is mandatory. C1-INH concentrate has been
their home refrigerator and their family doctor needs to
shown to be effective and save children with acute
be well informed about the disease. Moreover, parents
attacks and for short-term prophylaxis as well. Man-
and patients need to carry medical information cards
agement of long-term prophylaxis is difficult. Both
with information about the diagnosis in different
antifibrinolytic agents and attenuated androgens cannot
be recommended, although both are used where C1-INH concentrate is not available. Therefore, the devel-opment of new drugs is particularly important for
Treatment of acute attacks
children, who need long-term prophylactic pharmaco-
Acute HAE attacks are successfully treated by infusion of
10–30 units C1-INH concentrate/kg body weight, usually500–1000 units. If this is not effective within 30–60 min,another 500–1000 units are to be administered. This
Future directions – new treatment options
treatment is usually effective within <2 h, save and welltolerated. Any swelling in the head-and-neck area,
Significant adverse reactions, an uncomfortable route of
unexplained hoarseness or dyspnoea or acute abdominal
administration (i.v. infusion) and the lack of a proven
pain should be treated with C1-INH promptly.
preventive pharmacotherapy may affect the quality of lifeof patients with HAE. These call for new options to treatHAE as also AAE and RAE. Fortunately, there are three
Short-term prophylaxis
new pharmacotherapeutic options which are currently
Short-term prophylaxis is achieved by the infusion of
investigated in clinical trials (Fig. 8): (1) recombinant
500–1000 units within 30–60 min before a planned inter-
C1-INH, (2) the kallikrein inhibitor ecallantide and (3)
vention. It should be done in any surgical procedure,
the BKR-2-antagonist icatibant.
dental intervention or trauma in the head-and-neckregion.
Recombinant human C1-INH (rhC1-INH; Pharming
Long-term prophylaxis
Group, Leiden, the Netherlands) is extracted from the
In children, systematic data from large cohorts and
raw milk of transgenic rabbits. The gene-encoding
controlled or randomized studies are lacking. There are
human C1-INH is functionally linked to an extra bovine
no established criteria when to initiate long-term pro-
milk-specific promotor sequence (alpha-S1 casein) in the
phylaxis. Once it is initiated, it is very difficult to
rabbit DNA, thus leading to the secretion of C1-INH
discontinue and is then usually given lifelong. As
into the milk. rhC1-INH is expected to act in HAE the
C1-INH is a plasma product and expensive, it is not
same way as native C1-INH: it inhibits factor XIIa and
In addition, a prolongation of aPTT was observed. Thisis not surprising, as kallikrein and the Hagemann factor(factor XIIa), which autoactivate one another, are initialplayers in the clotting cascade. This action of ecallantidemight be a matter of concern and might cause significantinteractions with commonly used drugs, such as aspirinor heparin. However, it was suggested that prolongationof aPTT by ecallantide is not likely to represent anincreased risk of bleeding (126). Another concern even-tually limiting the use of ecallantide is the formation ofantibodies as well as the occurrence of anaphylacticreaction against this protein. Both events were observedduring treatment with ecallantide in a HAE patient, andmay rise safety concerns in terms of hypersensitivityreactions (127).
Target for new drugs to treat bradykinin-induced
kallikrein, and blocks the formation of bradykinin
Icatibant (Jerini AG, Berlin, Germany) is a synthetic
(Fig. 8). Nevertheless, the half-life of rhC1-INH is
decapeptide (MW: 1304.6 Da) with a similar structure
approximately 3 h which is much shorter than the
than bradykinin, but with five nonproteinogenic amino
half-life of native C1-INH (125). rhC1-INH is currently
acids. It is a potent, specific and selective BKR-2
tested in phase III trials on humans as listed in the web
antagonist that does not interact with other peptide
register of the U.S. National Institute of Health (http://
receptors, e.g. angiotensin II, substance P and neurokinin
www.clinicaltrials.gov; accessed 16 April 2007). In con-
A (128). Intranasal single-dose administration of icatib-
trast to the plasma pool C1-INH, which is still the
ant, at doses up to 500 lg and prior to kinin challenge,
therapy of choice, the use of rhC1-INH will reduce the
proved to inhibit bradykinin-induced symptoms in a
small but significant risk for the transmission of diseases
dose-dependent manner (129). Icatibant has been admin-
elicited by contaminations of human plasma with so far
istered so far to more than 1350 subjects in phase I–III
unknown pathogens, such as viruses and prions. The
trials (A. G. Jerini, personal communication). In an open-
virtual unlimited availability of recombinant proteins
label, proof-of-concept, phase II study, icatibant was
such as rhC1-INH may also reduce the still immense
administered either i.v. (0.4–0.8 mg/kg) or s.c. (30–40 mg)
costs for C1-INH therapy.
to 15 HAE patients with 20 attacks (130) . Symptomintensity significantly decreased within 4 h after theadministration of icatibant, and a recent case report of
treatment of HAE with icatibant supports these results
Ecallantide (DX-88, Dyax Corp., Cambridge, MA,
USA) is a recombinant 60 amino acid protein that
Icatibant caused no treatment-related serious adverse
specifically inhibits the plasma kallikrein. By blocking
events or withdrawals. Local rapidly disappearing
kallikrein, the formation of bradykinin from its precur-
cutaneous reactions (erythema) occur following s.c.
sor high-molecular kininogen is downregulated (Fig. 7),
injection (131). However, the upcoming results of two
suggesting that ecallantide roughly mimicks the physio-
double-blind, randomized phase III studies (FAST 1
logical actions of C1-INH. However, there are many
and 2) have to be awaited before the safety profile of
differences to the biological activity of C1-INH. For
icatibant can be further determined. In these studies,
example, although ecallantide potently inhibits plasma
the efficacy of a single s.c. dose (30 mg) of icatibant in
kallikrein, it shows a much less inhibitory activity
moderate-to-severe HAE attacks of the skin or the
against C1r, C1s, plasmin, factor XIIa and factor XIa
abdomen was tested. The primary efficacy end point
(126). Ecallantide has been tested in several phase I and
was the time to the onset of symptom relief assessed by
phase II trials, and is currently in a phase III trial for
the patient using a visual analogue scale. The secondary
treating HAE. In a phase II trial in 48 patients with
end points of the double-blind, either placebo-con-
HAE administered i.v. DX-88 significantly improved
trolled (FAST 1) or tranexamic acid-controlled (FAST
symptoms compared to placebo (126). The primary end
2), randomized multicentre study included the response
point was the proportion of patients reporting a signi-
rate (the rate of onset of symptom relief within 4 h
ficant improvement 4 h after the administration. Overall,
after application), the time to the complete alleviation
ecallantide is described to be well tolerated. Some
of symptoms, and the safety and tolerability of treat-
reported side effects were dizziness, fatigue, headache,
ment. The results of these recently finished studies have
nausea, vomiting and elevations of the liver function test.
not yet been published.
Nonallergic angioedema
1. Bas M, Hoffmann TK, Kojda G.
15. Sharma JN. Does kinin mediate the
29. Menke JG, Borkowski JA, Bierilo KK,
Evaluation and management of angi-
hypotensive action of angiotensin con-
MacNeil T, Derrick AW, Schneck KA
oedema of the head and neck. Curr
verting enzyme (ACE) inhibitors? Gen
et al. Expression cloning of a human
Opin Otolaryngol Head Neck Surg
B1 bradykinin receptor. J Biol Chem
16. Clements J, Hooper J, Dong Y, Harvey
2. Agostoni A, Cicardi M. Drug-induced
T. The expanded human kallikrein
30. Ma JX, Wang DZ, Ward DC, Chen L,
angioedema without urticaria. Drug
(KLK) gene family: genomic organisa-
Dessai T, Chao J et al. Structure and
tion, tissue-specific expression and
chromosomal localization of the gene
3. Zuberbier T. Urticaria. Allergy 2003;58:
potential functions. Biol Chem 2001;
(BDKRB2) encoding human bradyki-
nin B2 receptor. Genomics
4. Morwood K, Gillis D, Smith W, Kette
17. Britos J, Nolly H. Kinin-forming en-
F. Aspirin-sensitive asthma. Intern
zyme of rat cardiac tissue. Subcellular
31. Chai KX, Ni A, Wang D, Ward DC,
Med J 2005;35:240–246.
distribution and biochemical proper-
Chao J, Chao L. Genomic DNA se-
5. Kaplan AP, Greaves MW. Angioede-
ties. Hypertension 1981;3:II-5.
quence, expression, and chromosomal
ma. J Am Acad Dermatol 2005;53:
18. Sharma JN, Kesavarao U. Cardiac
localization of the human B1 bradyki-
kallikrein in hypertensive and normo-
nin receptor gene BDKRB1. Genomics
6. Alsenz J, Bork K, Loos M. Autoanti-
tensive rats with and without diabetes.
body-mediated acquired deficiency of
32. Regoli DC, Marceau F, Lavigne J.
C1 inhibitor. N Engl J Med 1987;316:
Induction of beta 1-receptors for kinins
19. Sharma JN, Uma K, Yusof AP. Left
in the rabbit by a bacterial lipopoly-
7. Agostoni A, Cicardi M. Hereditary and
ventricular hypertrophy and its relation
saccharide. Eur J Pharmacol
acquired C1-inhibitor deficiency: bio-
to the cardiac kinin-forming system in
logical and clinical characteristics in
hypertensive and diabetic rats. Int J
33. Bhoola KD, Figueroa CD, Worthy K.
235 patients. Medicine (Baltimore)
Bioregulation of kinins: kallikreins,
20. Nustad K, Vaaje K, Pierce JV. Syn-
kininogens, and kininases. Pharmacol
8. Messerli FH, Nussberger J. Vasopept-
thesis of kallikreins by rat kidney slices.
Rev 1992;44:1–80.
idase inhibition and angio-oedema.
Br J Pharmacol 1975;53:229–234.
34. Busse R, Fleming I. Regulation of
21. Yang HY, Erdos EG, Levin Y.
endothelium-derived vasoactive auta-
9. Agostoni A, ygoren-Pursun E, Binkley
A dipeptidyl carboxypeptidase that
coid production by hemodynamic for-
KE, Blanch A, Bork K, Bouillet L
converts angiotensin I and inactivates
ces. Trends Pharmacol Sci 2003;24:
et al. Hereditary and acquired angi-
bradykinin. Biochim Biophys Acta
oedema: problems and progress: pro-
35. Venema VJ, Ju H, Sun J, Eaton DC,
ceedings of the third C1 esterase
22. Yang HY, Erdos EG, Levin Y. Char-
Marrero MB, Venema RC. Bradykinin
inhibitor deficiency workshop and
acterization of a dipeptide hydrolase
stimulates the tyrosine phosphoryla-
beyond. J Allergy Clin Immunol
(kininase II: angiotensin I converting
tion and bradykinin B2 receptor
enzyme). J Pharmacol Exp Ther
association of phospholipase C gamma
10. Gainer JV, Nadeau JH, Ryder D,
1 in vascular endothelial cells. Biochem
Brown NJ. Increased sensitivity to
23. Vavrek RJ, Stewart JM. Competitive
Biophys Res Commun 1998;246:70–75.
bradykinin among African Americans.
antagonists of bradykinin. Peptides
36. Fleming I, Busse R. Tyrosine phos-
J Allergy Clin Immunol 1996;98:
phorylation and bradykinin-induced
24. Roberts RA. Bradykinin receptors:
signaling in endothelial cells. Am J
11. McDowell SE, Coleman JJ, Ferner RE.
characterization, distribution and
Systematic review and meta-analysis of
mechanisms of signal transduction.
37. Adams DJ, Barakeh J, Laskey R, Van
ethnic differences in risks of adverse
Prog Growth Factor Res 1989;1:
BC. Ion channels and regulation of
reactions to drugs used in cardiovas-
intracellular calcium in vascular
cular medicine. BMJ 2006;332:
25. Regoli D, Rhaleb NE, Drapeau G,
endothelial cells. FASEB J 1989;3:
Dion S. Kinin receptor subtypes.
12. Malde B, Regalado J, Greenberger PA.
J Cardiovasc Pharmacol 1990;15
38. Martin TW, Wysolmerski RB. Ca2+-
Investigation of angioedema associated
dependent and Ca2+-independent
with the use of angiotensin-converting
26. Regoli D, Barabe J. Pharmacology of
pathways for release of arachidonic
enzyme inhibitors and angiotensin
bradykinin and related kinins. Phar-
acid from phosphatidylinositol in
receptor blockers. Ann Allergy Asthma
macol Rev 1980;32:1–46.
endothelial cells. J Biol Chem
27. Hess JF, Borkowski JA, Young GS,
13. Pfeffer MA, McMurray JJ, Velazquez
Strader CD, Ransom RW. Cloning and
39. Ju H, Venema VJ, Liang H, Harris
EJ, Rouleau JL, Kober L, Maggioni
pharmacological characterization of a
MB, Zou R, Venema RC. Bradykinin
AP et al. Valsartan, captopril, or both
human bradykinin (BK-2) receptor.
activates the Janus-activated kinase/
in myocardial infarction complicated
Biochem Biophys Res Commun
signal transducers and activators of
by heart failure, left ventricular
transcription (JAK/STAT) pathway in
dysfunction, or both. N Engl J Med
28. McEachern AE, Shelton ER, Bhakta S,
vascular endothelial cells: localization
Obernolte R, Bach C, Zuppan P et al.
of JAK/STAT signalling proteins in
14. Abelous J, Bardier E. Les substance
Expression cloning of a rat B2 bra-
plasmalemmal caveolae. Biochem J
hypotensive de lu´rine humaine nor-
dykinin receptor. Proc Natl Acad Sci
male. CR Soc Biol 1909;66:511–512.
U S A 1991;88:7724–7728.
40. Bae SW, Kim HS, Cha YN, Park YS,
52. Mombouli JV, Nakashima M, Hamra
63. Alfie ME, Yang XP, Hess F, Carretero
Jo SA, Jo I. Rapid increase in endot-
M, Vanhoutte PM. Endothelium-
OA. Salt-sensitive hypertension in
helial nitric oxide production by bra-
dependent relaxation and hyperpolari-
bradykinin B2 receptor knockout mice.
dykinin is mediated by protein kinase
zation evoked by bradykinin in canine
Biochem Biophys Res Commun
A signaling pathway. Biochem Biophys
coronary arteries: enhancement by
Res Commun 2003;306:981–987.
exercise-training. Br J Pharmacol
64. Madeddu P, Varoni MV, Palomba D,
41. Madeddu P, Emanueli C, Gaspa L,
Emanueli C, Demontis MP, Glorioso
Salis B, Milia AF, Chao L et al. Role
53. Smith D, Gilbert M, Owen WG. Tissue
N et al. Cardiovascular phenotype of a
of the bradykinin B2 receptor in the
plasminogen activator release in vivo in
mouse strain with disruption of bra-
maturation of blood pressure pheno-
response to vasoactive agents. Blood
dykinin B2-receptor gene. Circulation
type: lesson from transgenic and
knockout mice. Immunopharmacology
54. Giannella E, Mochmann HC, Levi R.
65. Marceau F, Hess JF, Bachvarov DR.
Ischemic preconditioning prevents the
The B1 receptors for kinins. Pharmacol
42. Han ED, MacFarlane RC, Mulligan
impairment of hypoxic coronary vaso-
AN, Scafidi J, Davis AE III. Increased
dilatation caused by ischemia/reper-
66. Feletou M, Bonnardel E, Canet E.
vascular permeability in C1 inhibitor-
fusion: role of adenosine A1/A3 and
Bradykinin and changes in microvas-
deficient mice mediated by the bra-
bradykinin B2 receptor activation.
cular permeability in the hamster cheek
dykinin type 2 receptor. J Clin Invest
Circ Res 1997;81:415–422.
pouch: role of nitric oxide. Br J Phar-
55. Zhu P, Zaugg CE, Simper D,
43. Groves P, Kurz S, Just H, Drexler H.
Hornstein P, Allegrini PR, Buser PT.
67. Campos MM, Souza GE, Calixto JB.
Role of endogenous bradykinin in
Bradykinin improves postischaemic
In vivo B1 kinin-receptor upregulation.
human coronary vasomotor control.
recovery in the rat heart: role of high
Evidence for involvement of protein
energy phosphates, nitric oxide, and
kinases and nuclear factor kappaB
44. Hall JM. Bradykinin receptors: phar-
prostacyclin. Cardiovasc Res
pathways. Br J Pharmacol 1999;
macological properties and biological
roles. Pharmacol Ther 1992;56:
56. Leesar MA, Stoddard MF,
68. Dray A. Kinins and their receptors in
Manchikalapudi S, Bolli R. Bradyki-
hyperalgesia. Can J Physiol Pharmacol
45. Ellis EF, Heizer ML, Hambrecht GS,
nin-induced preconditioning in patients
Holt SA, Stewart JM, Vavrek RJ.
undergoing coronary angioplasty.
69. Lerner UH. Regulation of bone meta-
Inhibition of bradykinin- and kallik-
J Am Coll Cardiol 1999;34:639–650.
bolism by the kallikrein-kinin system,
rein-induced cerebral arteriolar dilation
57. Ritchie RH, Marsh JD, Lancaster WD,
the coagulation cascade, and the acute-
by a specific bradykinin antagonist.
Diglio CA, Schiebinger RJ. Bradykinin
phase reactants. Oral Surg Oral Med
blocks angiotensin II-induced hyper-
Oral Pathol 1994;78:481–493.
46. Marceau F, Regoli D. Bradykinin
trophy in the presence of endothelial
70. Cruwys SC, Garrett NE, Perkins MN,
receptor ligands: therapeutic perspec-
cells. Hypertension 1998;31:39–44.
Blake DR, Kidd BL. The role of bra-
tives. Nat Rev Drug Discov 2004;3:
58. Maestri R, Milia AF, Salis MB,
dykinin B1 receptors in the mainten-
Graiani G, Lagrasta C, Monica M
ance of intra-articular plasma
47. Duchene J, Schanstra JP, Pecher C,
et al. Cardiac hypertrophy and micro-
extravasation in chronic antigen-in-
Pizard A, Susini C, Esteve JP et al. A
vascular deficit in kinin B2 receptor
duced arthritis. Br J Pharmacol
novel protein-protein interaction be-
knockout mice. Hypertension
tween a G protein-coupled receptor
71. Couture R, Harrisson M, Vianna RM,
and the phosphatase SHP-2 is involved
59. Fuller RW, Dixon CM, Cuss FM,
Cloutier F. Kinin receptors in pain and
in bradykinin-induced inhibition of cell
Barnes PJ. Bradykinin-induced bron-
inflammation. Eur J Pharmacol
proliferation. J Biol Chem
choconstriction in humans. Mode of
action. Am Rev Respir Dis 1987;
72. Wang C, Chao L, Chao J. Direct gene
48. Hong SL. Effect of bradykinin and
delivery of human tissue kallikrein
thrombin on prostacyclin synthesis in
60. Tsukagoshi H, Sun J, Kwon O, Barnes
reduces blood pressure in spontane-
endothelial cells from calf and pig
PJ, Chung KF. Role of neutral
ously hypertensive rats. J Clin Invest
aorta and human umbilical cord vein.
endopeptidase in bronchial hyperre-
Thromb Res 1980;18:787–795.
sponsiveness to bradykinin induced by
73. Pesquero JB, Araujo RC, Heppenstall
49. Cocks TM, Angus JA, Campbell JH,
IL-1 beta. J Appl Physiol 1995;78:
PA, Stucky CL, Silva JA Jr, Walther T
Campbell GR. Release and properties
et al. Hypoalgesia and altered inflam-
of endothelium-derived relaxing factor
61. Mukae S, Aoki S, Itoh S, Iwata T,
matory responses in mice lacking kinin
(EDRF) from endothelial cells in cul-
Ueda H, Katagiri T. Bradykinin B(2)
B1 receptors. Proc Natl Acad Sci
ture. J Cell Physiol 1985;123:310–320.
receptor gene polymorphism is associ-
U S A 2000;97:8140–8145.
50. Palmer RMJ, Ferrige AG, Moncada S.
ated with angiotensin-converting en-
74. Ni A, Yin H, Agata J, Yang Z, Chao L,
Nitric oxide release accounts for the
zyme inhibitor-related cough.
Chao J. Overexpression of kinin B1
biological activity of endothelium-de-
receptors induces hypertensive re-
rived relaxing factor. Nature 1987;
62. Hulsmann AR, Raatgeep HR, Saxena
sponse to des-Arg9-bradykinin and
PR, Kerrebijn KF, de Jongste JC. Bra-
susceptibility to inflammation. J Biol
51. Gryglewski RJ, Palmer RM, Moncada
dykinin-induced contraction of human
S. Superoxide anion is involved in the
peripheral airways mediated by both
75. Yang C, Hsu WH. Glucose-depend-
breakdown of endothelium-derived
bradykinin beta 2 and thromboxane
ency of bradykinin-induced insulin
vascular relaxing factor. Nature
prostanoid receptors. Am J Respir Crit
secretion from the perfused rat pan-
Care Med 1994;150:1012–1018.
creas. Regul Pept 1997;71:23–28.
Nonallergic angioedema
76. Damas J, Bourdon V, Lefebvre PJ.
88. Danielsson A, Nilsson TK, Uddenfeldt
100. Hellebrand MC, Kojda G, Hoffmann
Insulin sensitivity, clearance and re-
P. Alterations in C1 inhibitor and
TK, Bas M. Angioedema due to ACE
lease in kininogen-deficient rats. Exp
clotting factor concentrations in pri-
inhibitors and AT(1) receptor antago-
mary biliary cirrhosis and other chro-
nists. Hautarzt 2005;57:808–810.
77. Duka I, Shenouda S, Johns C,
nic liver diseases. Scand J Gastro-
101. Jessup M, Brozena S. Heart failure.
Kintsurashvili E, Gavras I, Gavras H.
N Engl J Med 2003;348:2007–2018.
Role of the B(2) receptor of bradykinin
89. Cohen H, Hunt JB, Dixit M, Kanwar
102. Casas JP, Chua W, Loukogeorgakis S,
in insulin sensitivity. Hypertension
S, Thomas HC. Decreased contact
Vallance P, Smeeth L, Hingorani AD
factor mediated fibrinolysis in cirrho-
et al. Effect of inhibitors of the renin-
78. Rett K, Wicklmayr M, Dietze GJ.
sis. Br J Haematol 1993;85:542–545.
angiotensin system and other antihy-
Metabolic effects of kinins: historical
90. Gaur S, Cooley J, Aish L, Weinstein R.
pertensive drugs on renal outcomes:
and recent developments. J Cardiovasc
systematic review and meta-analysis.
Pharmacol 1990;15(Suppl. 6):S57–S59.
angioedema with normal C1-inhibitor
79. Kishi K, Muromoto N, Nakaya Y,
activity: does danazol work? Am J
103. Kaplan NM, Opie LH. Controversies
Miyata I, Hagi A, Hayashi H et al.
in hypertension. Lancet 2006;367:
Bradykinin directly triggers GLUT4
91. Monnier N, Ponard D, Duponchel C,
translocation via an insulin-independ-
Csopaki F, Bouillet L, Tosi M et al.
104. Bork K, Fischer B, Dewald G. Recur-
ent pathway. Diabetes 1998;47:
Characterisation of a new C1 inhibitor
rent episodes of skin angioedema and
mutant in a patient with hepatocellular
severe attacks of abdominal pain in-
80. Yusuf S, Sleight P, Pogue J, Bosch J,
carcinoma. Mol Immunol
duced by oral contraceptives or hor-
Davies R, Dagenais G. Effects of an
mone replacement therapy. Am J Med
92. Lefebvre J, Murphey LJ, Hartert TV,
itor, ramipril, on cardiovascular events
Jiao SR, Simmons WH, Brown NJ.
105. Bouillet L, Ponard D, Drouet C,
in high-risk patients. The Heart Out-
Dipeptidyl peptidase IV activity in pa-
Jullien D, Massot C. Angioedema and
comes Prevention Evaluation Study
tients with ACE-inhibitor-associated
oral contraception. Dermatology
Investigators. N Engl J Med 2000;
angioedema. Hypertension 2002;39:
106. Pichler WJ, Lehner R, Spath PJ.
81. Goring HD, Bork K, Spath PJ, Bauer
93. Dicpinigaitis PV. Angiotensin-convert-
Recurrent angioedema associated with
R, Ziemer A, Hintner H et al. Hered-
ing enzyme inhibitor-induced cough:
hypogonadism or anti-androgen ther-
itary angioedema in the German-
ACCP evidence-based clinical practice
apy. Ann Allergy 1989;63:301–305.
speaking region. Hautarzt 1998;49:
guidelines. Chest 2006;129:169S–173S.
107. Visy B, Fust G, Varga L, Szendei G,
94. Gainer JV, Morrow JD, Loveland A,
Takacs E, Karadi I et al. Sex hormones
82. Cicardi M, Zingale L, Zanichelli A,
King DJ, Brown NJ. Effect of bra-
in hereditary angioneurotic oedema.
Pappalardo E, Cicardi B. C1 inhibitor:
dykinin-receptor blockade on the re-
Clin Endocrinol (Oxf) 2004;60: 508–515.
molecular and clinical aspects. Springer
sponse to angiotensin-converting-
108. Agostoni A, Cicardi M, Cugno M,
Semin Immunopathol 2005; 27:
enzyme inhibitor in normotensive and
Zingale LC, Gioffre D, Nussberger J.
hypertensive subjects. N Engl J Med
Angioedema due to angiotensin-con-
83. Joseph K, Kaplan AP. Formation of
verting enzyme inhibitors. Immuno-
bradykinin: a major contributor to the
95. Nussberger J, Cugno M, Amstutz C,
innate inflammatory response.
Cicardi M, Pellacani A, Agostoni A.
109. Berkun Y, Shalit M. Hereditary angi-
Adv Immunol 2005;86:159–208.
Plasma bradykinin in angio-oedema.
oedema first apparent in the ninth
84. Bowen T, Cicardi M, Farkas H, Bork
decade during treatment with ACE
K, Kreuz W, Zingale L et al. Canadian
96. Kostis JB, Packer M, Black HR,
inhibitor. Ann Allergy Asthma Immu-
2003 International Consensus Algo-
Schmieder R, Henry D, Levy E. Om-
rithm For the Diagnosis, Therapy, and
apatrilat and enalapril in patients with
110. Bas M, Kojda G, Bier H, Hoffmann
Management of Hereditary Angioede-
hypertension: the Omapatrilat Cardio-
TK. Durch ACE-Hemmer induziertes
ma. J Allergy Clin Immunol 2004;
vascular Treatment vs. Enalapril
Angioo¨dem des Kopf-Hals-Bereichs.
(OCTAVE) trial. Am J Hypertens
Eine Frage der Zeit? HNO 2004;52:
85. Blanch A, Roche O, Urrutia I,
Gamboa P, Fontan G, Lopez-Trascasa
97. Campbell DJ, Krum H, Esler MD.
111. Bas M, Hoffmann TK, Bier H, Kojda
M. First case of homozygous C1
Losartan increases bradykinin levels in
G. Increased C-reactive protein in
inhibitor deficiency. J Allergy Clin
hypertensive humans. Circulation
Br J Clin Pharmacol 2005;59:233–238.
86. Cicardi M, Zingale LC, Pappalardo E,
98. Goodfriend TL, Elliott ME, Catt KJ.
112. Davis AE III. The pathophysiology of
Folcioni A, Agostoni A. Autoantibod-
Angiotensin receptors and their
hereditary angioedema. Clin Immunol
ies and lymphoproliferative diseases in
antagonists. N Engl J Med 1996;334:
acquired C1-inhibitor deficiencies.
113. Gompels MM, Lock RJ, Abinun M,
Medicine (Baltimore) 2003;82:274–281.
99. Gohlke P, Pees C, Unger T. AT2
Bethune CA, Davies G, Grattan C
87. Markovic SN, Inwards DJ, Frigas EA,
receptor stimulation increases aortic
et al. C1 inhibitor deficiency: consen-
Phyliky RP. Acquired C1 esterase
cyclic GMP in SHRSP by a kinin-
sus document. Clin Exp Immunol
inhibitor deficiency. Ann Intern Med
dependent mechanism. Hypertension
114. Shiber JR. Angioedema of the aryte-
noids. N Engl J Med 2005;353:e15.
115. Kojda G, Hafner D, Behne M,
122. Bork K, Pitton M, Harten P, Koch P.
127. Caballero T, Lopez-Serrano C. Ana-
Wilhelm M. Pharmakologie
Hepatocellular adenomas in patients
phylactic reaction and antibodies to
Toxikologie Systematisch, 2nd edn.
taking danazol for hereditary angio-
DX-88 (kallikrein inhibitor) in a patient
Bremen, London, Boston: UNI-MED
oedema. Lancet 1999;353:1066–1067.
with hereditary angioedema. J Allergy
123. Cicardi M, Zingale LC, Bergamaschini
Clin Immunol 2006; 117:476–477.
116. Farkas H, Harmat G, Fust G, Varga L,
L, Agostoni A. Angioedema associated
128. Icatibant. HOE 140, JE 049, JE049.
Visy B. Clinical management of her-
with angiotensin-converting enzyme
Drugs R D 2005;6:239–244.
editary angio-oedema in children.
inhibitor use: outcome after switching
129. Proud D, Bathon JM, Togias AG,
Pediatr Allergy Immunol 2002;13: 153–
to a different treatment. Arch Intern
Naclerio RM. Inhibition of the re-
sponse to nasal provocation with bra-
117. Taparia M, Cordingley FT, Leahy MF.
124. Bas M, Kojda G. Angioo¨dem nach
dykinin by HOE-140: efficacy and
Pulmonary embolism associated with
Einnahme von Irbesartan [Angioedema
duration of action. Can J Physiol
tranexamic acid in severe acquired
induced by irbesartan]. Apotheken-
haemophilia. Eur J Haematol
130. Rosenkranz B, Bork K, Frank J.
125. van Doorn MB, Burggraaf J, van Dam
Proof-of-concept study of icatibant
118. Dunn CJ, Goa KL. Tranexamic acid:
T, Eerenberg A, Levi M, Hack CE
(JE 049), a bradykinin B2 receptor
a review of its use in surgery and other
et al. A phase I study of recombinant
antagonist in treatment of hereditary
indications. Drugs 1999;57:1005–1032.
human C1 inhibitor in asymptomatic
angioedema. Clin Pharmacol Ther
119. Bucca C. Take the side-effects of drugs
patients with hereditary angioedema.
2005; 77:2 (Abstract).
into account. Lancet 2004;364:1285.
J Allergy Clin Immunol 2005;116:
131. Bas M, Bier H, Greve J, Kojda G,
120. Howes LG, Tran D. Can angiotensin
Hoffmann TK. Novel pharmacothera-
receptor antagonists be used safely in
126. Levy JH, O!Donnell PS. The thera-
py of acute hereditary angioedema with
patients with previous ACE inhibitor-
peutic potential of a kallikrein inhibitor
bradykinin B-2-receptor antagonist
induced angioedema? Drug Saf 2002;
for treating hereditary angioedema.
icatibant. Allergy 2006;61:1490–1492.
Expert Opin Investig Drugs 2006;
121. Gelfand JA, Sherins RJ, Alling DW,
Frank MM. Treatment of hereditaryangioedema with danazol. Reversal ofclinical and biochemical abnormalities.
N Engl J Med 1976;295:1444–1448.
Source: https://moodle.med.lu.se/pluginfile.php/27793/mod_book/chapter/5487/Nonallergic_angioedema_role_of_bradykinin.pdf
Evolution, 56(7), 2002, pp. 1331–1339 WITHIN- AND BETWEEN-POPULATION VARIATION FOR WOLBACHIA-INDUCED REPRODUCTIVE INCOMPATIBILITY IN A HAPLODIPLOID MITE F. VALA,1,2 A. WEEKS,3 D. CLAESSEN,4 J. A. J. BREEUWER,5 AND M. W. SABELIS6 Institute for Biodiversity and Ecosystem Dynamics, University of Amsterdam, P.O. Box 94084, 1090 GB Amsterdam, The Netherlands
NUEVAS FORMAS DE CREAR julio-agosto de 2004 julio-agosto de 2004 Revista de la OMPI/ Revista de la OMPI/ A primera vista, el olor del césped particular de un producto pueden Marcas tridimensionales y Las empresas deben prestar especial recién cortado, el color lila, el grito de adquirir un carácter distintivo y con- formas de productos atención a la descripción y a la repre-