Neurolipidomics.org

Apoptosis (2007) 12:969–977DOI 10.1007/s10495-007-0755-3
HIV protease inhibitors modulate apoptosis signaling in vitro and
in vivo
Stacey R. Vlahakis · Steffany A. L. Bennett ·
Shawn N. Whitehead · Andrew D. Badley
Published online: 9 February 2007
� Springer Science + Business Media, LLC 2007
Abstract HIV protease inhibitors are an integral part of
Keywords Apoptosis . HIV protease inhibitors . Neurons .
effective anti-HIV therapy. The drugs block HIV protease,
prevent proper packaging of HIV virions, and decrease theHIV viral burden in the peripheral blood of infected individ-
HIV infection causes a progressive depletion of CD4+ T
uals. In addition to direct anti-viral effects, the HIV protease
cells. There are likely many reasons for the CD4+ T cell
inhibitors also modulate apoptosis. A growing body of work
loss, including apoptosis. Current therapy for HIV-infected
demonstrates the anti-apoptotic effects of HIV protease in-
patients often includes a combination of reverse transcriptase
hibitors on CD4+ and CD8+ T cells during HIV infection.
inhibitors and HIV protease inhibitors. The first HIV protease
The mechanism of this apoptosis inhibition is supported by
inhibitor was FDA approved for anti-HIV therapy in humans
several proposed hypotheses for how they alter the fate of
in 1995. Currently, there are ten HIV protease inhibitors that
the cell, including preventing adenine nucleotide translo-
are approved for clinical use in humans (saquinavir, indi-
cator pore function, which consequently prevents loss of
navir, darunavir, nelfinavir, lopinavir, amprenavir, atazanavir,
mitochondrial transmembrane potential. More recently, the
tipranavir, ritonavir, and fosamprenavir). These drugs are in-
anti-apoptotic effects of the HIV protease inhibitors have
hibitors of the HIV viral aspartyl protease. The compounds
been tested in non-HIV, non-immune cell, whereby protease
have a strong affinity for the active site of the HIV pro-
inhibitors prevent apoptosis, and disease in animal models of
tease and irreversibly inhibit the catalytic activity of the
sepsis, hepatitis, pancreatitis and stroke. Interestingly, when
HIV protease inhibitors are used at supra-therapeutic con-
When HIV protease is inhibited, viral particles are pro-
centrations, they exert pro-apoptotic effects. This has been
duced, but they are immature, and do not package prop-
demonstrated in a number of tumor models. Although it is
erly into infectious virions. Moreover, naturally occurring
unclear how HIV protease inhibitors can induce apoptosis
HIV protease mutations, which arise during suboptimal an-
at increased concentrations, future research will define the
tiretroviral therapy, result in impaired replication kinetics
targets of the immunomodulation and reveal the full clinical
of progeny virions. In addition to its role in viral replica-
potential of this intriguing class of drugs.
tion, HIV protease may also contribute to HIV pathogenesis.
When transfected into a human or bacterial cell, HIV pro-tease is cytotoxic and causes cleavage of a variety of hostproteins including actin, Bcl2 and procaspase 8. It remainsunclear how HIV protease initiates cell death.
S. R. Vlahakis · A. D. Badley (�)
Although the HIV protease inhibitors have limited
Division Infectious Disease, Mayo Clinic College of Medicine,200 First Street SW, Rochester, MN 55905
bioavailability and stability, they are the basis of effec-
tive anti-HIV therapy, and in combination with other an-tiretroviral agents, produce a sustained decrease in HIV viral
S. A. L. Bennett · S. N. Whitehead
load. Over the past ten years, evidence has accumulated that
Neural Regeneration Laboratory, Department of Biochemistry,Microbiology and Immunology, University of Ottawa,
HIV protease inhibitors have non-viral effects on the host
451 Smyth Road, Ottawa, ON, Canada, K1M 1E5
cells beyond the effect of blocking HIV protease enzymatic

Apoptosis (2007) 12:969–977
activity. For example, HIV protease inhibitors also directly
significantly when a patient is treated with HIV PI containing
affect cellular apoptosis signaling.
HAART, which correlates with a decrease in HIV peripheralblood viral load and an increase in peripheral blood CD4+T cells. In addition, CD4+ and CD8+ T cells isolated from
The role of HIV protease inhibitors during treatment of
HIV-infected individuals before and on days 1, 4, and 8,
after starting treatment with HIV PI containing HAART, haddecreased sensitivity to Fas-mediated apoptosis after being
Optimal anti-HIV therapy is called highly active antiretrovi-
on HAART for as little as one day
ral therapy (HAART). This includes the combination of HIV
A recent trial by Landay et al. focused on the mecha-
reverse transcriptase inhibitors, often with an HIV protease
nism behind HIV protease inhibitor immune effects. The
inhibitor. Since the advent of HAART in late 1995, these reg-
trial compared patients with suppressed HIV viral replica-
imens have been shown to increase CD4+ T cell counts and
tion on a PI-based anti-HIV regimen or non-PI based drug
reduce the amount of HIV virus quantitated in the peripheral
regimen. One week after enrollment in the study, subjects in
blood, in infected individuals. However, in many cases, the
the PI containing arm had less spontaneous T cell apoptosis
increase in CD4+ T cells that occurs during HIV therapy
than those in the non-PI containing arm. Although there are
appeared to result from actions that were independent of the
likely multiple reasons for CD4+ T cell decline during the
effect of the drug on viral replication. In one such instance,
course of HIV infection, these data suggest that patients on
HIV-infected subjects were randomized in a clinical trial to
HIV protease inhibitors have less spontaneous CD4+ T cell
receive therapy with three drugs, including one protease in-
apoptosis and have improved CD4+ T cell counts, further
hibitor (saquinivir, zidovudine, and zalitabine) or two drugs.
suggesting that the HIV protease inhibitors block CD4+ T
One of the two drug regimens included a protease inhibitor
cell apoptosis, independent of their effect on HIV replication
(zidovudine and saquinivir), and the other two-drug regimen
had no protease inhibitors (zidovudine and zalitabine) Two-hundred and eighty subjects completed 24 weeks oftreatment. Even though there was worse virologic control in
In vitro anti-apoptotic properties of HIV protease
the two-drug, than the three-drug regimen group, the sub-
jects in the two-drug group who received the HIV PI hadimproved CD4+ T cell counts, compared to the two-drug
Due to findings that PI therapy reduced lymphocyte apopto-
group that did not receive an HIV PI. Subsequent clinical tri-
sis, our group proposed in 1998 that highly active antiretro-
als confirmed that HIV protease inhibitors improved CD4+
viral therapy (HAART) might independently block cellu-
T cell counts in HIV-infected individuals, independent of the
lar apoptosis Subsequent investigations tested and con-
viral suppression In a meta-analysis comparing anti-
firmed this finding ex vivo and in vitro HIV PIs
HIV drug regimens using a PI to those that switched the PI
anti-apoptotic effects were investigated using ritonavir in
to a non-nucleoside reverse transcriptase inhibitor (NNRTI),
cultures of bone marrow cells from HIV-infected patients or
PI-based therapy resulted in higher CD4+ T cell counts
normal controls. Hematopoietic colony forming unit replica-
The analysis required that both PI and non-PI based regi-
tion, following addition of ritonavir, had 45% less apoptosis
mens have complete HIV viral suppression. Therefore, the
than untreated cultures. The authors also reported a decrease
CD4+ T cell benefit with HIV protease inhibitor therapy ap-
in caspase-1 expression after ritonavir treatment
peared to be, in part, unrelated to the effect on HIV viral
In additional experiments investigating the effects of HIV
suppression. In a different trial comparing treatment with
PIs on T cell death during HIV infection, CD4+ T cells
HIV protease containing highly active antiretroviral therapy
isolated from healthy uninfected individuals had increased
(HAART) to protease inhibitor therapy alone, there was less
Fas expression and Fas and anti-CD3-induced apoptosis
virologic suppression of the subjects on PI mono-therapy.
when incubated with HIV virions. Inducible Fas expres-
However, there was no decrease in CD4+ T cell counts over
sion and apoptosis were abrogated when the cells were pre-
one year, even in the group who received PI mono-therapy
incubated with the HIV PI, saquinavir These findings
and virologically failed Additionally, in a comparison be-
were supported by subsequent investigations which reported
tween PI-based anti-HIV therapy to NNRTI-based anti-HIV
that saquinavir and ritonavir inhibited TNF-mediated U937
therapy, there was equal viral suppression in both groups;
cell apoptosis in a dose-dependent fashion with 38–60% re-
however, PI-based regimens had a greater increase in CD4+
duction in apoptosis in PI treated cells During HIV
infection, the HIV envelope gp120 binds to the CD4 and
In the absence of effective antiretroviral therapy, HIV-
CXCR4 receptors on the surface of the CD4+ T cell and
infected subjects have considerable lymphocyte apoptosis in
signals the cell to undergo apoptosis. This bystander death
lymphoid tissue. The lymphoid tissue apoptosis is reduced
is one of the ways that HIV depletes the immune system.

Apoptosis (2007) 12:969–977
Matarrese et al. treated human CD4+ T cells with HIV
were expanded in a study that demonstrated that caspases-3,
gp120 to make the cell sensitive to Fas-mediated apopto-
-6, and -8 activity was not inhibited by indinavir in U937 cells
sis, which resulted in mitochondrial changes and apoptosis
at drug concentrations that effectively inhibited U937 apop-
after Fas exposure. When the CD4+ T cells were pretreated
tosis Nelfinavir did not block activation of caspases-1,
with a PI before HIV gp120, mitochondrial depolorization
-3, -4, -5, -9, and -8 in lysates of Jurkat T cells undergoing
was blocked in a whole cell or cell-free system, or isolated
Fas-mediated apoptosis
mitochondria Taken together, this data indicates that
In addition to caspases, the calpain proteases have been
patients who received HIV protease inhibitors had improved
considered as a possible site for HIV PIs to mediate apopto-
CD4+ T cell counts independent of the state of HIV viral
sis. Calpains are Ca+-dependent cysteine proteases reported
replication, and in vitro work confirmed that HIV PIs can
to be involved in several models of apoptosis, including U937
inhibit T cell apoptosis, specifically that induced by HIV.
cells, but are not absolutely required for apoptosis Be-cause HIV PIs are designed to inhibit the HIV cysteine pro-tease, they may influence cellular apoptosis by blocking cal-
Mechanism of HIV protease inhibitors apoptosis
pain activation and function. Ghibelli et al. demonstrated in
a U937 model of apoptosis that indinavir and ritonavir di-rectly inhibit apoptosis in cell systems where calpains are
The mechanism of HIV protease inhibitor-mediated apopto-
activated and block m-calpain activation Other inves-
sis inhibition is being actively investigated (Table HIV PIs
tigators have demonstrated that ritonavir competitively in-
inhibit the proteolytic activity of HIV viral protease, there-
hibited activity of both m- and -µ calpain isoforms in PC12
fore, it has been postulated that they might have a similar
cells Other investigators could not confirm this ob-
effect on other cellular proteases. Together with the obser-
servation. These authors postulate that concentrations of ri-
vations that HIV PIs block Fas-mediated apoptosis, many
tonavir close to the maximum solubility of the drug, which
investigators hypothesize that HIV PIs may inhibit the cas-
was used in previous reports, may have artificially altered
pase family members and block apoptosis. Although this is
the results A calpain inhibitor could have significant
an attractive model, caspases are cystine proteases and HIV
therapeutic implications beyond HIV apoptosis, including
protease is an aspartyle protease. Several groups have inves-
neurodegenerative diseases; however, the evidence remains
tigated the direct effect of HIV PIs on intracellular caspase
unclear whether HIV PIs have a significant effect on calpain
activity. When recombinant caspases-1, -3, -6, -7, or -8 were
incubated with a fluorogenic tetrapeptide substrate for each
An alternative model suggests that HIV protease in-
caspase in the presence of absence of nelfinavir, the HIV
hibitors alter the expression of apoptotic regulatory pro-
PI inhibited HIV protease cleavage of gag/pol, but did not
teins. Although early studies reported a change in Fas ex-
inhibit the activity of any of the caspases These results
pression after PI treatment, subsequent work did not show a
Possible methods for HIV protease inhibitor regulating apoptosis
Theoretical site of action
HIV PIs block Fas and TNF mediated apoptosisHIV PIs do not block caspase-1, -3, -4, -5, -6, -7, -8, or -9 activity in
HIV PIs inhibit apoptosis in cell systems where calpains are activatedHIV PIs blocked m-calpain activation in U937 cellsHIV PIs inhibited activity of both m- and -µ calpain isoforms in PC12
HIV PI did not inhibit m- or µ-calpain hydrolysis or activation at lower,
Apoptosis regulatory proteins
No change in Fas protein levels after HIV PI treatmentNo change in RNA levels of Fas, Fas L, and TNF after HIV PI treatmentNo change in Bcl-2, Bax, and Bcl-XL after HIV PI treatment
Mitochondrial transmembrane potential
HIV PIs maintain mitochondrial membrane integrity after apoptosis
HIV PIs prevent cytochrome c release from mitochondria after
apoptosis stimuli
ANT (adenine nucleotide translocator) necessary for HIV PI to block
mitochondria mediated apoptosis

Apoptosis (2007) 12:969–977
change in Fas levels with HIV PI therapy Bone
release a fluorescent dye after pore opening, did not result
marrow progenitor cells from HIV-infected individuals in-
in pore opening when pretreated with nelfinavir before the
cubated with ritonavir and indinavir showed no change in
RNA levels of Fas, or Fas L. Two reports have specifically
In summary, there are several theories regarding the mech-
investigated the levels of intracellular apoptotic regulatory
anism by which HIV PIs inhibit cellular apoptosis. The most
proteins with and without HIV PI treatment. Protein levels
data suggest that HIV PIs block apoptosis by maintaining
of Bcl2, Bax, and Bcl-XL were evaluated by flow cytom-
mitochondrial integrity, likely by HIV PIs preventing pore
etry, and were unchanged after PI administration
function of the adenine nucleotide translocator subunit of the
Therefore, HIV PIs can block Fas- and TNF-mediated apop-
mitochondrial permeability transition pore complex (Fig.
tosis; this effect does not appear to be due to changes inintra- or extracellular apoptotic regulatory protein levels
HIV protease inhibitor anti-apoptotic properties during
HIV PIs may also block apoptosis at the level of the
non-HIV disease states
mitochondria by disrupting the transmitochondrial mem-brane potential. The mitochondrial transmembrane potential
The novel anti-apoptotic properties of HIV PIs are being
occurs from an asymmetric distribution of ions on both sides
evaluated in preclinical studies as a potential therapy for dis-
of the inner mitochondrial membrane that is maintained
ease states associated with increased levels of apoptosis such
by the mitochrondrial permeability transition pore complex
as sepsis, the leading cause of death in critically ill patients.
(PTPC). After an apoptotic signal, the PTPC opens,
The original description of in vivo use of HIV PIs for non-
disrupts the membrane potential, and releases apoptogenic
HIV related disease was in a mouse sepsis model. Sepsis is
factors, including cytochrome c and procaspase-9. Thus, the
the leading cause of death in critically ill patients. Despite
mitochondria serves as a regulatory checkpoint of apoptotic
advances in supportive care, mortality from sepsis remains
signaling. Many regulatory proteins, including Bcl2 family
high. Animal studies demonstrate that sepsis results in ex-
members and IAPs, interact at the level of the mitochondria
tensive lymphocyte apoptosis, as well as intestinal epithelial
to alter apoptosis. In the first report of the effects of HIV
cell apoptosis These findings have been confirmed
PI on mitochondrial integrity, Jurkat cell Fas-mediated
during autopsy studies in humans who died of sepsis
apoptosis was inhibited with 10 µM of nelfinavir, a dose
In a mouse model of sepsis, created by cecal ligation
that would simulate physiologic levels if taken clinically.
and perforation, mice pretreated with HIV PIs had improved
Nelfinavir treated cells maintained intact mitochondrial
survival and reduced lymphocyte apoptosis The HIV
transmembrane potential, as determined by DioC6 staining,
PI treated mice had an increase in the Th1 cytokine TNFα
a lipophilic dye that stains the mitochondria The
and a reduction in the TH2 cytokines IL-6 and IL-10. It ap-
authors also reported that 10 µM of nelfinavir inhibited cy-
pears the beneficial effect of PI treatment was due to reduced
tochrome c release during Fas-induced apoptosis. The HIV
lymphocyte apoptosis because lymphocyte deficient Rag 1
accessory molecule, Vpr, caused PTPC opening and loss of
–/– mice had no benefit from HIV PI treatment. Follow-up
transmitochondrial potential when added directly to mito-
work by Weaver et al. investigated the effect of HIV PIs
chondria. Nelfinavir pretreatment of Jurkat cells prevented
during Staphylococcal enterotoxin B+DGal-induced shock.
Vpr-induced DioC6 release from the mitochondria and
There was a 60% improvement in 24-h survival in mice pre-
cell death. Other groups have confirmed that HIV protease
treated with HIV PIs than those treated with vehicle control.
inhibitors block mitochondrial transmembrane potential loss
In addition, the authors also demonstrated that HIV PI pre-
in multiple models of apoptosis In a recent
treatment reduced mouse death from Fas-induced fatal hep-
report, Weaver et al. investigated how HIV PIs influence
atitis and middle cerebral artery occlusion-induced stroke
mitochondrial integrity by using yeast models. Wild type or
yeast deficient in voltage-dependent anion channel (VDAC)or adenine nucleotide translocator (ANT) isoforms, twocomponents of the PTPC, were treated with Vpr or H2O2
HIV protease inhibitors and stroke
which induce mitochondrial apoptosis. Apoptosis onlyoccurred after Vpr or H2O2 treatment when ANT was
Cerebral ischemia or stroke occurs when blood flow (and
present. Furthermore, when Jurkat cells were pretreated
thus oxygen) to the brain is reduced through hemorrhage
with nelfinavir and then an agonist for VDAC, there was
or clot-induced occlusion of blood vessels. The only ther-
no inhibition of mitochondrial potential loss or apoptosis.
apy with proven clinical benefit is thrombolysis requiring
However, 10 µM of nelfinavir blocked ANT agonist-induced
administration of tissue plasminogen activator within 3 h of
mitochondrial transmembrane potential loss and apoptosis
the onset of ischemic attack and/or oral aspirin within the first
Lastly, proteoliposomes reconstituted with ANT, which
48 h after stroke onset Although the pathophysiology
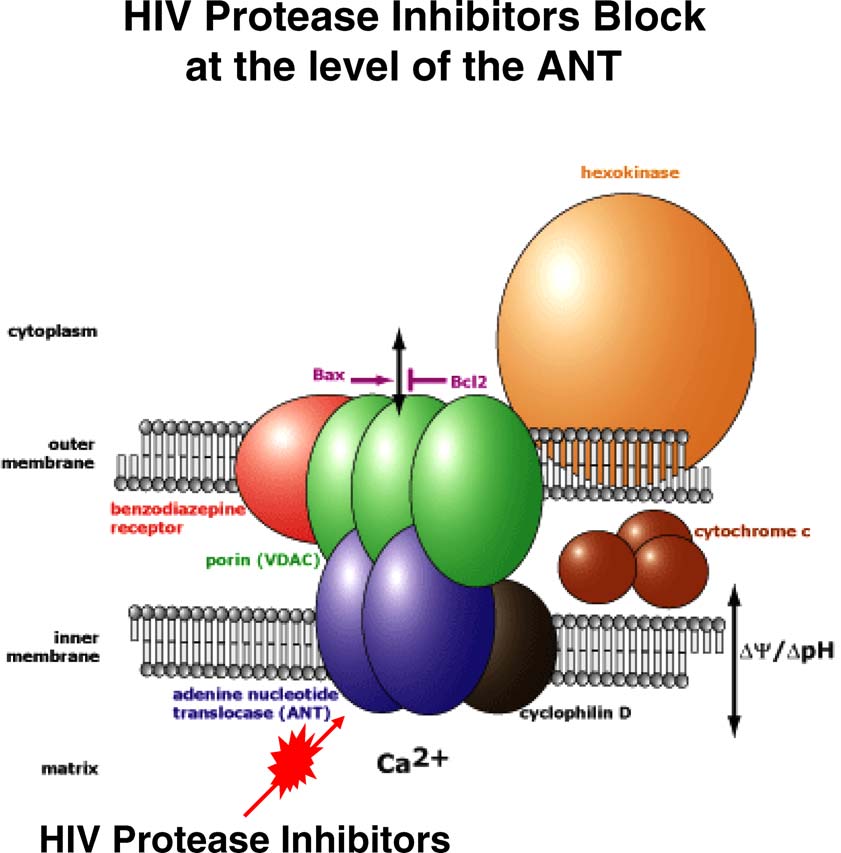

Apoptosis (2007) 12:969–977
Fig. 1 HIV protease inhibitors block cellular apoptosis at the level of the adenine nucleotide translocase (ANT) in the mitochondrial pore complex.
The figure has been adapted from the Mitosciences Inc. website
of stroke damage extends well beyond this time frame, no
Na+, Cl−, and Ca2+ ions. Neuronal depolarization resulting
clinical intervention capable of targeting discrete molecular
from a loss of calcium homeostasis is further exacerbated
mechanisms of cell death has been shown to effectively pro-
by the sustained release of glutamate and reduction in ex-
tect neurons from subsequent neuronal loss. Elucidation of
tracellular pH leading to excitotoxicity. Production of free
the complex and multiple cell death pathways initiated by
fatty acids and oxyradicals are elevated, triggering oxida-
ischemia has made it increasingly apparent that a broader
tive remodeling of membrane lipids, impaired glial home-
therapeutic approach is required These findings are
ostatic functions and enhanced inflammatory cell activation
supported by correlative clinical evidence of neuroprotective
activity in other central nervous system disorders following
Studies using primary hippocampal neurons indicate that
treatment with PIs
HIV PIs can protect neurons from cell death triggered by
Stroke elicits rapid necrotic and excitotoxic mechanisms
membrane lipid remodeling associated with oxidative stress.
as well as delayed apoptotic-like responses. Cell death is
Ritonavir has been shown to inhibit neuronal injury triggered
initiated once core tissue is deprived of oxygen-rich blood.
in vitro by 4-hydroxynonenal, a lipid-soluble aldehydic prod-
Immediately following vessel occlusion, cell viability is lost,
uct of membrane peroxidation that impairs Na+, K+, and -
in part, by a reduction in ATP levels resulting in an efflux of
ATPase activity In vivo, administration of nelfinavir and
K+ ions from compromised neurons and glia and an influx of
ritonavir prior to ischemic insult effectively reduces infarct

Apoptosis (2007) 12:969–977
size in mice following middle cerebral artery occlusion, re-sulting in functional recovery of ischemic neurons
Mechanistic assessment suggests that HIV PIs act to re-
duce delayed neuronal ischemic death triggered also throughmitochondrial-dependent pathways. Neurons located at theperiphery of the necrotic core (dubbed the penumbra) arespared acute ischemic injury. Damage is not evident untilhours, days, and weeks following reperfusion when cells be-gin to exhibit many of the morphological and biochemicalcharacteristics of apoptosis Death is likely triggeredduring ischemia/reperfusion by downstream release of in-ternal calcium stores from endoplasmic reticulum and mito-chondria Additional death pathways are subsequentlyinitiated by a complex cross-talk between extrinsic deathreceptor mediated-induction and intrinsic mitochondrial-dependent pathways. Briefly, extrinsic induction involvesbinding of apoptogens, such as Fas ligand and tumor necro-sis factor α (TNFα), to death domain-containing receptorsinducing oligomerization and recruitment of the adapter pro-teins FADD and TRADD Complex formation betweenadapter proteins, receptor death domains, and procaspasesaccelerates the autocatalytic activation of procaspase-8, -10,and -2. Once cleaved, these initiator caspases can cleave andactivate executioner caspases-3, -6, and -7 responsible for theregulated disassembly of cellular proteins characteristic ofapoptosis Intrinsic cell death is dictated by mitochon-drial function. Release of reactive oxygen species followingischemic reperfusion triggers release of cytochrome c andATP from compromised mitochondria into the cytosol, oftenwith concomitant loss of loss of �ym. Activation of caspase9 occurs with formation of the apoptosome composed ofAPAF-1, cytochrome c, ATP, and procaspase 9 resulting in
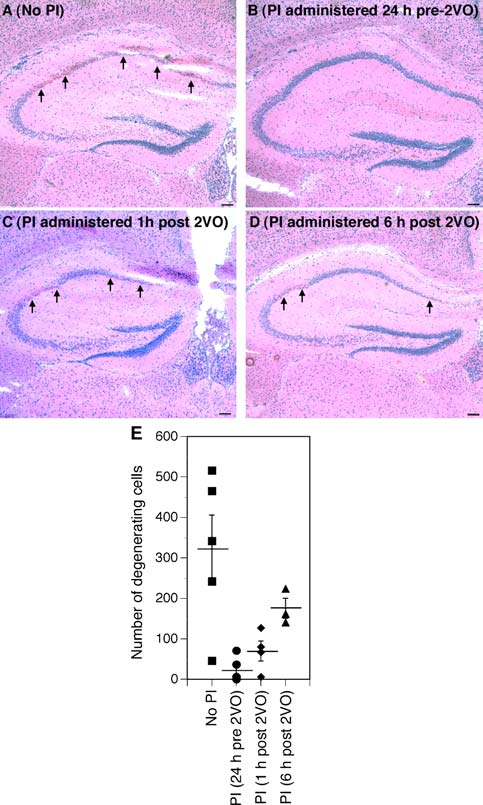
Effects of nelfinavir and ritonavir on ischemic hippocampal
downstream activation of caspase-3, -6, -7
damage induced by two vessel carotid occlusion. Mice subjected to
HIV PIs act at multiple mitochondrial branch points of
two vessel occlusion (A) were compared to animals that received three
these overlapping apoptotic cascades to reduce delayed neu-
oral gavages of either nelfinavir or ritonavir 8 h apart starting 24 h
ronal death following ischemic insult. Pretreatment with nel-
before (B), 1 h after (C), or 6 h after (D) carotid occlusion. Arrowsindicate areas of ischemic damage. Viable cell number in the CA1 was
finavir prior to apoptotic challenge inhibits the pore function
counted 48 h after ischemic onset in hemotoxylin and eosin-stained
of the adenine nucleotide transporter, thereby reducing the
sections (E). Data represent the average number of viable cells in
loss of loss of �y
both hippocampi and are expressed per animal. The mean number of
m, apoptosome formation, and downstream
viable cells per condition ± standard error of measurement is indi-
caspase activation Because HIV PIs inhibit the down-
cated. Neurodegeneration was completely attenuated by pre-treatment
stream mitochondrial events elicited by caspase 8-mediaed
with PIs and partially attenuated by administration of PIs following
cleavage of Bid, downstream events of the extrinsic pathway
carotid occlusion. Scale bars, 100 µm
are also reduced
Recent unpublished data from our laboratories indicate
that this protection may also be of potential clinical benefit
mals received nelfinavir boosted by ritonavir prior to or im-
even if administered after the ischemic event—a key require-
mediately after 2VO surgery (Fig. Neuroprotection was
ment for successful adjuvant therapies. Loss of CA1 pyra-
still evident, albeit at reduced efficacy, when nelfinavir and
midal neurons in the hippocampus is a hallmark of global
ritonavir were administered up to 6 h after two-vessel carotid
ischemia. Ischemic hippocampal injury can be experimen-
occlusion (Fig. suggesting a wider therapeutic window
tally designed by the two-vessel occlusion rodent model of
than previously anticipated
global ischemia in which both internal carotid arteries are
While promising, mechanistic assessment of HIV PIs
transiently ligated. As demonstrated in Fig. we observed
in stroke management is still in early days. Although
significant sparing of CA1 pyramidal neurons whether ani-
HIV PIs appear to inhibit apoptosis in multiple organs at
Apoptosis (2007) 12:969–977
concentrations comparable to that seen in the plasma follow-
drug may interfere with cellular activation signals that result
ing clinical application (reviewed in these same com-
in transformation and protect the tumor cell from apopto-
pounds trigger cell death at higher concentrations
sis. High concentrations of ritonavir (10–50 µM) inhibit the
Moreover, chronic administration, as part of HIV treatment,
proliferation of murine and human tumor cell lines. DNA
has been associated with an enhanced risk of ischemia in
laddering demonstrated that 15–30 µM of ritonavir induced
both central and peripheral tissue. Chronic HIV PI treatment
apoptosis in the same cell lines Of note, the concen-
can elicit hyperlipidemia, accelerating atherosclerosis and
trations of ritonavir used were over 15 times what most
represent a potential increased risk for myocardial infarction
adults achieve at FDA-approved doses and at which the
As such, it is unlikely that HIV PIs can be used
drug blocks apoptosis. This led to further work developing
prophylactically to prevent stroke damage in susceptible in-
the pro-apoptotic effects of HIV PIs. Adult T-cell leukemia
dividuals. However, the promise of transient administration
(ATL) is an aggressive malignancy associated with human
is promising. The impact of acute administration of HIV
T-cell leukemia virus (HTLV) and is very resistant to con-
PIs following stroke requires a thorough evaluation of the
ventional chemotherapy. When ATL cells were incubated
therapeutic window open to mitochondrial manipulation and
with 20–40 µM of ritonavir, there was a five-fold increase
is essential to validate the promise of HIV PIs as a poten-
in spontaneous apoptosis, resulting in a similar decrease in
tial adjuvant strategy to promote neuronal survival following
tumor cell survival In ATL cell lines and primary ATL
cells 40 µM of ritonavir inhibited transcriptional activationof NF-κB. In addition, HIV PIs inhibited the expression ofthe targets of NF-κB, Bcl-XL, survivin, c-Myc, and cyclin D2
HIV protease inhibitors and pancreatitis
There is also evidence that HIV protease inhibitors are
Accelerated apoptosis also contributes to cellular injury dur-
pro-apoptotic in models of solid tumors. Freshly isolated
ing acute pancreatitis. Our group investigated whether treat-
multiple myeloma cells from patients under went apopto-
ment with the HIV protease inhibitors nelfinavir/ritonavir
sis after incubation with 40–50 µM of ritonavir, saquinavir,
would reduce the severity of pancreatitis using a mouse
and nelfinavir. This was associated with a decrease in
caerulein-induced pancreatitis model. Ritonavir was used to
the anti-apoptotic protein Mcl-1, and blocked IL-6 phos-
increase nelfinavir drug levels to a therapeutic level in the
phorylation of ERK 1/2 and STAT 3 Different tu-
mice. Mice treated with nelfinavir/ritonavir before caerulein
mor types may have different mechanisms by which HIV
induced pancreatitis had reduced serum amylase levels and
PIs inhibit tumor growth and promote apoptosis. Riton-
less acinar injury of the pancreas on histologic review, com-
avir used at 20 µM in solid and hematologic tumor models
pared to mice pretreated with vehicle control.
caused an increase in the cellular concentrations of the anti-proliferative and pro-apoptotic proteasome substrate cdk in-hibitor, p21. This was associated with a block in prote-
HIV protease inhibitors and T cell production
olytic degradation consistent with an earlier reportthat HIV PI impact proteasome activity Accumula-
The anti-apoptotic effects of HIV protease inhibitors may
tion of intracellular p21 resulted in cell-cycle arrest in G1
have indirect beneficial clinical effects as well. In a recent re-
phase and subsequent apoptosis in ritonavir treated tumor
port by Graham et al., five out of seven HIV-negative patients
treated with an HIV protease inhibitor containing HAART
HIV protease inhibitors are well-tolerated drugs with en-
regimen for a needle-stick exposure experienced a 3-log in-
ticing possibilities for future use to alter apoptosis in many
crease in thymic-derived na¨ıve T cells in the peripheral blood.
human disease states. Clinical trials of HIV PIs in disease
The increase in na¨ıve T cells, as determined by T cell recep-
states other than HIV are already underway investigating the
tor recombination excision circle levels (TREC), occurred
in vivo effects on human cellular apoptosis (NCT00346619
after four weeks of therapy, suggesting, in yet another set-
and NCT00233948). Furthermore, now that the target for
ting, that HIV PI can have beneficial immunemodulatory
how HIV PIs block cellular apoptosis has been identi-
fied as the mitochondrial pore protein ANT, physiochemi-cal optimization can be performed. Lastly, the mechanismby which high concentrations of HIV PIs induce apopto-
Paradoxical pro-apoptotic effect of HIV protease
sis in transformed tumor models needs to be further clari-
fied. Studies are needed to determine whether the HIV PIsalter proteins that modulate cellular proliferation or apop-
Under certain conditions HIV PIs may be pro-apoptotic as
tosis, and whether there is a threshold to the pro-apoptotic
well. At increased doses of HIV PI, it is possible that the
Apoptosis (2007) 12:969–977
SRV is supported by a grant from the Mayo
16. Matarrese P, Tinari A, Gambardella L et al (2005) HIV pro-
Program in Translational Immunovirology and Biodefense and a Robert
tease inhibitors prevent mitochondrial hyperpolarization and re-
and Arlene Kogod Program on Aging Award from the Mayo Foun-
dox imbalance and decrease endogenous uncoupler protein-2 ex-
dation. ADB is supported by NIH grants RO1-AI62261 and RO1-
pression in gp 120-activated human T lymphocytes. Antivir Ther
AI40384, and a Burroughs Wellcome Award ID#1005160.
10(Suppl 2):M29–M45
17. Phenix BN, Lum JJ, Nie Z, Sanchez-Dardon J, Badley AD. (2001)
Antiapoptotic mechanism of HIV protease inhibitors: preventing
mitochondrial transmembrane potential loss. Blood 98(4):1078–1085
18. Ghibelli L, Mengoni F, Lichtner M et al (2003) Anti-apoptotic
1. Collier AC, Coombs RW, Schoenfeld DA et al (1996) Treatment of
effect of HIV protease inhibitors via direct inhibition of calpain.
human immunodeficiency virus infection with saquinavir, zidovu-
Biochem Pharmacol 66(8):1505–1512
dine, and zalcitabine: AIDS Clinical Trials Group. N Engl J Med
19. Chavan S, Kodoth S, Pahwa R, Pahwa S (2001) The HIV pro-
tease inhibitor Indinavir inhibits cell-cycle progression in vitro in
2. Kravcik S, Magill A, Sanghvi B et al (2001) Comparative CD4
lymphocytes of HIV-infected and uninfected individuals. Blood
T-cell responses of reverse transcriptase inhibitor therapy with or
without nelfinavir matched for viral exposure. HIV Clin Trials
20. Spinedi A, Oliverio S, Di Sano F, Piacentini M (1998) Calpain in-
volvement in calphostin C-induced apoptosis. Biochem Pharmacol
3. Deeks SG, Grant RM. (1999) Sustained CD4 responses after vi-
rological failure of protease inhibitor-containing therapy. Antivir
21. Wan W, DePetrillo PB (2002) Ritonavir inhibition of calcium-
Ther 4(Suppl 3):7–11
activated neutral proteases. Biochem Pharmacol 63(8):1481–1484
4. Albrecht MA, Bosch RJ, Hammer SM et al (2001) Nelfinavir,
22. Cuerrier D, Nie Z, Badley AD, Davies PL (2005) Ritonavir
efavirenz, or both after the failure of nucleoside treatment of HIV
does not inhibit calpain in vitro. Biochem Biophys Res Commun
infection. N Engl J Med 345(6):398–407
5. Staszewski S, Morales-Ramirez J, Tashima KT et al (1999)
23. Sloand EM, Kumar PN, Kim S, Chaudhuri A, Weichold FF, Young
Efavirenz plus zidovudine and lamivudine, efavirenz plus indi-
NS (1999) Human immunodeficiency virus type 1 protease in-
navir, and indinavir plus zidovudine and lamivudine in the treat-
hibitor modulates activation of peripheral blood CD4(+) T cells
ment of HIV-1 infection in adults. Study 006 Team. N Engl J Med
and decreases their susceptibility to apoptosis in vitro and in vivo.
Blood 94(3):1021–1027
6. Owen C, Kazim F, Badley AD (2004) Effect on CD4 T-cell count
24. Lu W, Andrieu JM (2000) HIV protease inhibitors restore
of replacing protease inhibitors in patients with successful HIV
impaired T-cell proliferative response in vivo and in vitro:
suppression: a meta-analysis. Aids 18(4):693–695
a viral-suppression-independent mechanism. Blood 96(1):250–
7. Arribas JR, Pulido F, Delgado R et al (2005) Lopinavir/ritonavir as
single-drug therapy for maintenance of HIV-1 viral suppression:
25. Isgro A, Aiuti A, Mezzaroma I et al (2005) HIV type 1 protease
48-week results of a randomized, controlled, open-label, proof-of-
inhibitors enhance bone marrow progenitor cell activity in nor-
concept pilot clinical trial (OK Study). J Acquir Immune Defic
mal subjects and in HIV type 1-infected patients. AIDS Res Hum
Syndr 40(3):280–287
8. Fethi T, Asma J, Amine SM et al (2005) Effects on immunological
26. Matarrese P, Gambardella L, Cassone A, Vella S, Cauda R, Mal-
and virological outcome of patients using one protease inhibitor
orni W (2003) Mitochondrial membrane hyperpolarization hijacks
or one non-nucleoside reverse transcriptase inhibitor in a triple
activated T lymphocytes toward the apoptotic-prone phenotype:
antiretroviral therapy: normal clinical practice versus clinical trial
homeostatic mechanisms of HIV protease inhibitors. J Immunol
findings. Curr HIV Res 3(3):271–276
9. Badley AD, Dockrell DH, Algeciras A et al (1998) in vivo analysis
27. Weichold FF, Bryant JL, Pati S, Barabitskaya O, Gallo RC, Reitz Jr
of Fas/FasL interactions in HIV-infected patients. J Clin Invest
MS (1999) HIV-1 protease inhibitor ritonavir modulates suscepti-
bility to apoptosis of uninfected T cells. J Hum Virol 2(5):261–269
10. Landay AL, Spritzler J, Kessler H et al (2003) Immune recon-
stitution is comparable in antiretroviral-naive subjects after 1
Mitochondrion-mediated apoptosis in HIV-1 infection. Trends
year of successful therapy with a nucleoside reverse-transcriptase
Pharmacol Sci 24(6):298–305
inhibitor- or protease inhibitor-containing antiretroviral regimen. J
29. Garg H, Blumenthal R (2006) HIV gp41-induced apoptosis is
Infect Dis 188(10):1444–1454
mediated by caspase-3-dependent mitochondrial depolarization,
11. Badley AD, Parato K, Cameron DW et al (1999) Dynamic correla-
which is inhibited by HIV protease inhibitor nelfinavir. J Leukoc
tion of apoptosis and immune activation during treatment of HIV
Biol 79(2):351–362
infection. Cell Death Differ 6(5):420–432
30. Miro O, Villarroya J, Garrabou G et al (2005) in vivo effects of
12. Phenix BN, Angel JB, Mandy F et al (2000) Decreased HIV-
highly active antiretroviral therapies containing the protease in-
associated T cell apoptosis by HIV protease inhibitors. AIDS Res
hibitor nelfinavir on mitochondrially driven apoptosis. Antivir Ther
Hum Retroviruses 16(6):559–567
13. Sloand EM, Maciejewski J, Kumar P, Kim S, Chaudhuri A, Young
31. Weaver JG, Tarze A, Moffat TC et al (2005) Inhibition of ade-
N. (2000) Protease inhibitors stimulate hematopoiesis and de-
nine nucleotide translocator pore function and protection against
crease apoptosis and ICE expression in CD34(+) cells. Blood
apoptosis in vivo by an HIV protease inhibitor. J Clin Invest
14. Estaquier J, Lelievre JD, Petit F et al (2002) Effects of antiretroviral
32. Ayala A, Herdon CD, Lehman DL, Ayala CA, Chaudry IH (1996)
drugs on human immunodeficiency virus type 1-induced CD4(+)
Differential induction of apoptosis in lymphoid tissues during sep-
T-cell death. J Virol 76(12):5966–5973
sis: variation in onset, frequency, and the nature of the mediators.
15. Wolf T, Findhammer S, Nolte B, Helm EB, Brodt HR. (2003)
Blood 87(10):4261–4275
Inhibition of TNF-alpha mediated cell death by HIV-1 specific
33. Hotchkiss RS, Swanson PE, Cobb JP, Jacobson A, Buchman TG,
protease inhibitors. Eur J Med Res 8(1):17–24
Karl IE (1997) Apoptosis in lymphoid and parenchymal cells dur-
Apoptosis (2007) 12:969–977
ing sepsis: findings in normal and T- and B-cell-deficient mice.
49. Pajonk F, Himmelsbach J, Riess K, Sommer A, McBride WH
Crit Care Med 25(8):1298–1307
(2002) The human immunodeficiency virus (HIV)-1 protease in-
34. Husain KD, Coopersmith CM (2003) Role of intestinal epithelial
hibitor saquinavir inhibits proteasome function and causes apop-
apoptosis in survival. Curr Opin Crit Care 9(2):159–163
tosis and radiosensitization in non-HIV-associated human cancer
35. Coopersmith CM, Chang KC, Swanson PE et al (2002) Overex-
cells. Cancer Res 62(18):5230–5235
pression of Bcl-2 in the intestinal epithelium improves survival in
50. Zhong DS, Lu XH, Conklin BS et al (2002) HIV protease in-
septic mice. Crit Care Med 30(1):195–201
hibitor ritonavir induces cytotoxicity of human endothelial cells.
36. Weaver JG, Rouse MS, Steckelberg JM, Badley AD (2004) Im-
Arterioscler Thromb Vasc Biol 22(10):1560–1566
proved survival in experimental sepsis with an orally administered
51. Bode H, Lenzner L, Kraemer OH et al (2005) The HIV protease
inhibitor of apoptosis. Faseb J 18(11):1185–1191
inhibitors saquinavir, ritonavir, and nelfinavir induce apoptosis and
37. Ly JV, Zavala JA, Donnan GA (2006) Neuroprotection and throm-
decrease barrier function in human intestinal epithelial cells. An-
bolysis: combination therapy in acute ischaemic stroke. Expert
tivir Ther 10(5):645–655
Opin Pharmacother 7(12):1571–1581
52. Penzak SR, Chuck SK (2002) Management of protease inhibitor-
38. Lee JM, Zipfel GJ, Choi DW (1999) The changing landscape of
associated hyperlipidemia. Am J Cardiovasc Drugs 2(2):91–
ischaemic brain injury mechanisms. Nature 399(6738 Suppl):A7–
53. Lai S, Lai H, Celentano DD et al (2003) Factors associated with
39. Reed JC (2001) Apoptosis-regulating proteins as targets for drug
accelerated atherosclerosis in HIV-1-infected persons treated with
discovery. Trends Mol Med 7(7):314–319
protease inhibitors. AIDS Patient Care STDS 17(5):211–219
40. Lo EH, Moskowitz MA, Jacobs TP (2005) Exciting, radical, sui-
54. Cohen CJ (2005) Ritonavir-boosted protease inhibitors, Part 2:
cidal: how brain cells die after stroke. Stroke 36(2):189–192
cardiac implications of lipid alterations. AIDS Read 15(10):528–
41. Rosenfeldt V, Valerius NH, Paerregaard A (2000) Regression of
HIV-associated progressive encephalopathy of childhood during
55. Graham DB, Bell MP, Huntoon CJ et al (2005) Increased thymic
HAART. Scand J Infect Dis 32(5):571–574
output in HIV-negative patients after antiretroviral therapy. Aids
42. MacGowan DJ, Scelsa SN, Waldron M (2001) An ALS-like syn-
drome with new HIV infection and complete response to antiretro-
56. Gaedicke S, Firat-Geier E, Constantiniu O et al (2002) Antitumor
viral therapy. Neurology 57(6):1094–1097
effect of the human immunodeficiency virus protease inhibitor
43. Lopez-Neblina F, Toledo AH, Toledo-Pereyra LH (2005) Molecu-
ritonavir: induction of tumor-cell apoptosis associated with per-
lar biology of apoptosis in ischemia and reperfusion. J Invest Surg
turbation of proteasomal proteolysis. Cancer Res 62(23):6901–
44. MacDonald JF, Xiong ZG, Jackson MF (2006) Paradox of Ca2+
57. Dewan MZ, Uchihara JN, Terashima K et al (2006) Efficient inter-
signaling, cell death and stroke. Trends Neurosci 29(2):75–81
vention of growth and infiltration of primary adult T-cell leukemia
45. Wan W, DePetrillo PB (2002) Ritonavir protects hippocampal neu-
cells by an HIV protease inhibitor, ritonavir. Blood 107(2):716–
rons against oxidative stress-induced apoptosis. Neurotoxicology
58. Ikezoe T, Saito T, Bandobashi K, Yang Y, Koeffler HP, Taguchi H
46. Ferrer I, Planas AM (2003) Signaling of cell death and cell survival
(2004) HIV-1 protease inhibitor induces growth arrest and apop-
following focal cerebral ischemia: life and death struggle in the
tosis of human multiple myeloma cells via inactivation of signal
penumbra. J Neuropathol Exp Neurol 62(4):329–339
transducer and activator of transcription 3 and extracellular signal-
47. Zheng Z, Zhao H, Steinberg GK, Yenari MA (2003) Cellular and
regulated kinase 1/2. Mol Cancer Ther 3(4):473–479
molecular events underlying ischemia-induced neuronal apoptosis.
59. Andre P, Groettrup M, Klenerman P et al (1998) An inhibitor of
Drug News Perspect 16(8):497–503
HIV-1 protease modulates proteasome activity, antigen presenta-
48. Phenix BN, Cooper C, Owen C, Badley AD (2002) Modulation of
tion, and T cell responses. Proc Natl Acad Sci USA 95(22):13120–
apoptosis by HIV protease inhibitors. Apoptosis 7(4):295–312
Source: http://neurolipidomics.org/NRL%20publications/2007Valhakis.pdf
Examen : BEP Date de l'épreuve : Spécialité/option : Carrières sanitaires et sociales Repère de l'épreuve : EP2 Épreuve : Sciences et technologie (En majuscules, suivi s'il y a lieu du nom d'épouse) Prénoms : N° du candidat (le numéro est celui qui figure sur la convocation ou la
Aghssa et al. Reproductive Health (2015) 12:85 DOI 10.1186/s12978-015-0053-4 Optimal cutoff value of basal anti-mullerianhormone in iranian infertile women forprediction of ovarian hyper-stimulationsyndrome and poor response to stimulation Malek Mansour Aghssa1, Azam Manshadi Tarafdari1*, Ensieh Shahrokh Tehraninejad1, Mohammad Ezzati2,Maryam Bagheri1, Zahra Panahi1, Saeed Mahdavi3 and Mehrshad Abbasi4