Nutrient-sensitized screening for drugs that shift energy metabolism from mitochondrial respiration to glycolysis

Nutrient-sensitized screening for drugs that shift energy
metabolism from mitochondrial respiration to glycolysis
Vishal M Gohil1–3,7, Sunil A Sheth1–3,7, Roland Nilsson1–3, Andrew P Wojtovich4,5, Jeong Hyun Lee6,
Fabiana Perocchi1–3, William Chen1–3, Clary B Clish2, Cenk Ayata6, Paul S Brookes4,5 & Vamsi K Mootha1–3
Most cells have the inherent capacity to shift their reliance on glycolysis relative to oxidative metabolism, and studies in model
systems have shown that targeting such shifts may be useful in treating or preventing a variety of diseases ranging from cancer to
ischemic injury. However, we currently have a limited number of mechanistically distinct classes of drugs that alter the relative
activities of these two pathways. We screen for such compounds by scoring the ability of > 3,500 small molecules to selectively
impair growth and viability of human fibroblasts in media containing either galactose or glucose as the sole sugar source.
We identify several clinically used drugs never linked to energy metabolism, including the antiemetic meclizine, which attenuates
mitochondrial respiration through a mechanism distinct from that of canonical inhibitors. We further show that meclizine
pretreatment confers cardioprotection and neuroprotection against ischemia-reperfusion injury in murine models. Nutrient-sensitized
screening may provide a useful framework for understanding gene function and drug action within the context of energy metabolism.
Virtually all cells exhibit metabolic flexibility and are capable of shift-
media. Nutrient-sensitized screening is based on the evidence that
ing their reliance on glycolysis relative to mitochondrial respiration. mammalian cells redirect their energy metabolism in response to the
Such shifts can occur at different timescales through a variety of mech-
available sugar sourc. Culturing cells in galactose as the sole sugar
anisms, allowing cells to cope with prevailing nutrient availability or source forces mammalian cells to rely on mitochondrial oxidative
energetic demands. There is mounting evidence of the therapeutic phosphorylation (OXPHOS) and was previously used to diagnose
potential of targeting such shifts. For example, many cancer cells rely human mitochondrial disorders or drug toxicity15,16. By screening
on aerobic glycolysis (the Warburg effect) and a recent study has our chemical library for drugs that selectively inhibit cell growth and shown that pharmacological agents that shift their metabolism toward proliferation in galactose- relative to glucose-containing media, we
mitochondrial respiration can retard tumor growt. Conversely, identify several FDA-approved compounds that redirect oxidative studies in animal models have shown that attenuating mitochondrial metabolism to glycolysis. We pursue the mechanism and potential
respiration can prevent the pathological consequences of ischemia-
clinical utility of one drug, meclizine, which is available without
reperfusion injury in myocardial infarction and stroke3–7.
prescription, crosses the blood-brain barrier and has never, to our
These observations motivate the search for agents that can safely knowledge, been linked to energy metabolism.
induce shifts in cellular energy metabolism in humans. Promising
work in this area has focused on hypoxia-inducible factor (HIF) a RESULTS
well-studied transcriptional regulator of genes involved in the cel-
A metabolic state–dependent growth and viability assay
lular adaptation to hypoxia9,10. HIF inhibitors and activators have Consistent with previous studies focused on other cell types14,17, we been identified through both academic and pharmaceutical drug found that human MCH58 skin fibroblasts grown in glucose derive screens and exhibit preclinical efficacy in treating cancer and in ATP from both aerobic glycolysis and mitochondrial glutamine oxida-ischemic disease respectively. Other approaches to treat ischemic tion ). However, when these cells are grown in galactose they injury include induced hypothermia, which has met with mixed exhibit a five- to sixfold decrease in the extracellular acidification results. New classes of agents, that can be titrated to safely shift rate (ECAR), reflecting decreased glycolysis, and a twofold increase energy metabolism, may yet provide important therapeutic value in in the oxygen consumption rate (OCR), consistent with a switch to several human diseases.
glutamine oxidatio (. Moreover, cells grown in galactose-
Here we use a nutrient-sensitized screening strategy to identify drugs containing medium maximize mitochondrial ATP production by
that shift cellular energy metabolism based on their selective effect using a larger fraction of mitochondrial respiration for ATP synthesis
on cell growth and viability in glucose- versus galactose-containing (Supplementary Fig. 1).
1Center for Human Genetic Research, Massachusetts General Hospital, Boston, Massachusetts, USA. 2Broad Institute of Massachusetts Institute of Technology and Harvard, Cambridge, Massachusetts, USA. 3Department of Systems Biology, Harvard Medical School, Boston, Massachusetts, USA. 4Department of Pharmacology and Physiology and 5Department of Anesthesiology, University of Rochester Medical Center, Rochester, New York, USA. 6Stroke and Neurovascular Regulation Laboratory, Department of Radiology, Massachusetts General Hospital, Charlestown, Massachusetts, USA. 7These authors contributed equally to this work. Correspondence should be addressed to V.K.M. ([email protected]).
Received 23 November 2009; accepted 12 January 2010; published online 14 February 2010;
nature biotechnology VOLUME 28 NUMBER 3 MARCH 2010


Positive Sglu/gal scores indicate drugs that were
selectively lethal or inhibited growth in galac-
tose-containing medium, such as inhibitors
of OXPHOS. Negative Sglu/gal scores may arise
from inhibition of glycolysis or from inhibi-
tion of proliferation, as fibroblasts cultured in
glucose grow more rapidly (Supplementary
(mpH/min/30 k cells)
Fig. 3b). For most drugs, Sglu/gal is close to
zero, indicating similar effects on growth and
OCR (pmol/min/30 k cells)
viability in glucose- and galactose-containing
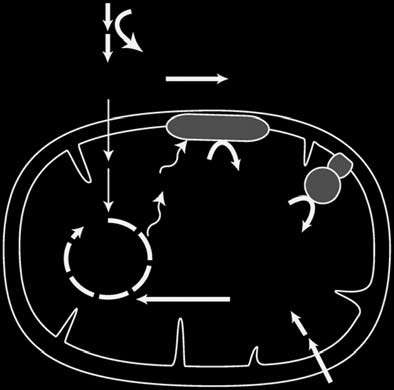
Figure 1 Metabolic plasticity of human fibroblasts. (a,b) Schematic representation of cellular
media (and Supplementary Table 1).
energy metabolism pathways. (a) Cells grown in glucose-rich medium derive ATP from glycolysis as
Reassuringly, the upper tail of the Sglu/gal
well as from glutamine-driven respiration. TCA, tricarboxylic acid; ETC, electron transport chain.
distribution ( is highly enriched
(b) Replacing glucose with galactose forces cells to generate ATP almost exclusively from glutamine-
for known respiratory chain and OXPHOS
driven oxidative metabolism. (c) Measurement of ECAR, a proxy for the rate of glycolysis, and
inhibitors: the top 25 compounds include
OCR, a proxy for mitochondrial respiration, of fibroblasts grown in media containing 10 mM glucose
20 compounds previously known to disrupt
or 10 mM galactose for 3 d. Data are expressed as mean ± s.d. (n = 5).
respiration by directly interrupting mito-chondrial respiration or uncoupling it from
The metabolic flexibility of fibroblasts permits screening for com-
ATP synthesis (Supplementary Table 2). Conversely, the lower tail is
pounds that retard growth or are lethal to cells only in a given metabolic enriched for known antineoplastic agents (): 14 of the 25 lowest
state. In a pilot experiment, we confirmed nutrient-dependent drug sen-
Sglu/gal scores correspond to known chemotherapeutic agents that are
sitivity of fibroblasts to known inhibitors of OXPHOS (Supplementary
likely to retard the growth and viability of cells rapidly proliferating
Fig. 2). To screen a library of chemicals, we designed a high-
in glucose (Supplementary Table 3).
throughput microscopy-based growth assay to identify compounds
We next asked if any clinically used drugs exhibit high Sglu/gal scores.
that differentially affect growth and viability in galactose- relative Among the top 2% of the Sglu/gal distribution (83 compounds), we
to glucose-containing media (and Supplementary Fig. 3a). identified 25 agents that have been used clinically (Supplementary
Because the proliferation rates are higher for cells provided with Table 4). Previous reports provide evidence that 9 out of these 25 drugs
glucose instead of galactose as their sole sugar source (Supplementary (papaverine, phenformin, artemisinin, pentamidine (NebuPent),
Fig. 3b), we considered the normalized cell number in each of the two clomiphene (Clomid), pimozide (Orap), niclosamide, fluvastatin
nutrient conditions. By measuring growth and survival over a 3-day (Lescol), carvedilol (Coreg)) can directly inhibit or uncouple the mito-
period, we were able to increase our ability to discover compounds chondrial respiratory chain (Supplementary Table 4). This list includes
with even subtle effects on energy metabolism.
two antimalarial drugs (mefloquine (Lariam) and artemisinin),
the latter of which has been reported to require mitochondrial res-
A small-molecule screen for agents that shift energy metabolism
piration in the malarial parasite for its action. The remaining 16
We screened a library of 3,695 chemical compounds in duplicate. clinically used agents cover a broad range of indications and diverse
The library has been previously describedand consists of two com-
mechanisms of action and, to our knowledge, have never been linked
mercially available compound collections that span nearly half of all to energy metabolism. We were particularly interested in identifying
FDA-approved drugs, as well as other bioactives and natural products. compounds that induce subtle metabolic shifts, as they may represent We analyzed the glucose and galactose results jointly to assign each particularly safe drugs with which to manipulate energy metabolism. drug a score, Sglu/gal, defined as the log ratio of the normalized cell To this end, we focused on commercially available drugs exhibiting
number in glucose divided by the normalized cell number in galac-
low to intermediate, positive Sglu/gal scores (0.15–0.45). We carried
tose. The full table of results is provided in Supplementary Table 1. out secondary assays of OCR, ECAR and cell viability and confirmed
a Glucose
OCR/ECAR (% of DMSO)
All data (n = 3,695)
Known OXPHOS inhibitors (n = 29)
Antineoplastic drugs (n = 140)
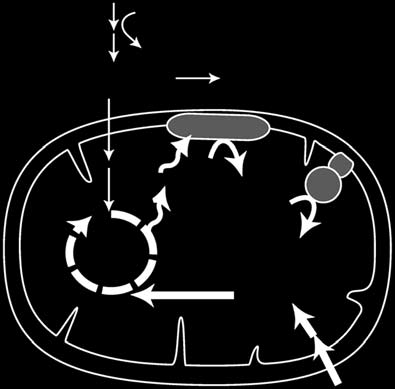
Figure 2 A nutrient-sensitized screen to discover agents that shift energy metabolism. (a) Schematic of the drug screen. MCH58 fibroblasts grown
in 96-well plates in glucose- or galactose-containing media are exposed to a chemical library of 3,695 compounds for 72 h. The logarithm of the
normalized cell number in glucose versus galactose provides a summary statistic (Sglu/gal) for each compound. (b) Results from a nutrient-sensitized
screen. Sglu/gal is plotted for top and bottom 250 compounds. (c) Secondary assays to evaluate compounds with modest yet positive Sglu/gal scores. The
OCR/ECAR ratio of selected compounds is plotted against the compounds' corresponding Sglu/gal score from b. OCR and ECAR measurements were made
on MCH58 fibroblasts grown in glucose and are normalized to cell viability. Compounds indicated by red symbols exhibited a statistically significant decrease in the OCR/ECAR ratio based on at least three independent replicates (P < 0.05; two-sided t-test).
VOLUME 28 NUMBER 3 MARCH 2010 nature biotechnology
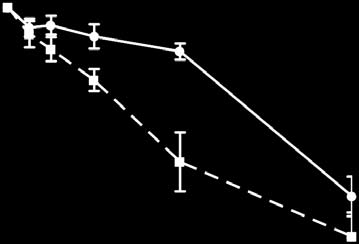
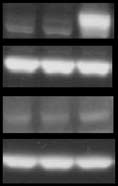

Figure 3 Effects of meclizine on cellular energy metabolism. (a) Cell
viability of MCH58 fibroblasts cultured in glucose- or galactose-containing
media with varying doses of meclizine for 3 d. Data are expressed as mean
± s.d. (n = 5). (b) OCR in MCH58 fibroblasts cultured in glucose-containing
medium with varying doses of meclizine for 200 min. Data are expressed as
mean ± s.d. (n = 3). (*, P < 0.05; **, P < 0.005; two-sided t-test). (c,d) OCR (c)
(pmol/min/30 k cells)
and ECAR (d) in multiple cell types cultured in glucose-containing medium
with 50 µM meclizine or DMSO for 200 min. Data are expressed as mean
± s.d. (n ≥ 3). (*, P < 0.05; two-sided t-test). (e) Time course of meclizine
(50 µM)-mediated OCR reduction over DMSO baseline compared to other
inhibitors of OXPHOS (1 µM each) in HEK293 cells. Data are expressed
as mean ± s.d. (n ≥ 3). (f) HIF-1α and HIF-2α detection by western blot
analysis of protein extract from HeLa cells after 6-h treatment with 0.1%
DMSO, 100 µM deferoxamine (DFO) or 50 µM meclizine. The complete
(pmol/min/30 k cells)
(mpH/min/30 k cells)
immunoblot is provided as Supplementary Figure 7b.
ECAR occurred in all cell types tested, including immortalized mouse
Meclizine (50 µM)
Antimycin (1 µM)
striatal cells, human embryonic kidney cells (HEK293) and HeLa
DFO (100 µM) – – +
cells ( and Supplementary Fig. 4). Although meclizine is
Meclizine (50 µM) – + –
classified as a histamine receptor (H
1) antagonist and a weak mus-
carinic acetylcholine receptor antagonist, the other 64 annotated
H1 receptor antagonists and 33 annotated antimuscarinic antagonists
(% of baseline) –80
in our chemical library did not exhibit elevated Sglu/gal scores (anti-
cholinergic P = 0.26, anti-H1 P = 0.77; Mann-Whitney rank sum test).
We tested two classic antihistamines—pyrilamine and pheniramine
that eight of these agents induce statistically significant (P < 0.05) as well as two well-characterized antimuscarinic agents—atropine metabolic shifts (. Of these eight clinically used drugs, we and scopolamine for their ability to inhibit OCR. Unlike meclizine,
were particularly interested in meclizine (Antivert), which has been these agents did not inhibit cellular OCR (Supplementary Fig. 5).
approved for the treatment of nausea and vertigo for decades, is These results suggest that meclizine's effect on energy metabo-
available over the counter, has a favorable safety profile, and likely lism occurs by means of a mechanism not involving cholinergic or
penetrates the blood-brain barrier given its efficacy in disorders of histamine receptors.
the central nervous syste.
Meclizine's suppression of cellular oxygen consumption occurred
with much slower kinetics than canonical inhibitors of OXPHOS that
Meclizine attenuates respiration in intact cells
directly target the respiratory chain or ATP synthase (. The slow
In secondary assays, we replicated our screening result and confirmed kinetics suggest that it takes time for meclizine to accumulate in mito-that galactose-grown cells are more sensitive to increasing doses chondria or alternatively, that it might act indirectly. To distinguish
of meclizine (. In agreement with our secondary screening between these alternatives, we studied the effect of meclizine on iso-assay (, treatment with meclizine reduced the OCR in a dose-
lated mitochondria. Using glutamate and malate, pyruvate and malate
dependent manner in cells cultured in glucose-rich medium ). or succinate as fuel substrates, we found no effect of meclizine on res-Meclizine-induced reduction in OCR and concomitant increase in piration of isolated mitochondria . Meclizine did not have
in solution (nmol)
in solution (nmol) 2
in solution (nmol) 25
e 0.40 Mito
Figure 4 Effect of meclizine on bioenergetics of isolated mitochondria. (a–c) Acute effect of meclizine
on oxygen consumption in isolated mitochondria using glutamate/malate (a), succinate (b) or pyruvate/
malate (c) as substrates. Traces are representative of five independent measurements. (d) Acute effect
of meclizine on mitochondrial membrane potential measured with tetramethyl rhodamine methyl ester
(TMRM) in isolated mitochondria. Traces are representative of five independent measurements. (e) Acute
effect of meclizine on mitochondrial NADH in isolated mitochondria. Traces are representative of five independent measurements. Mitochondria (Mito), glutamate and malate (G/M), succinate (S), pyruvate/
malate (P/M), meclizine (Mec) or DMSO, ADP and carbonyl cyanide m-chlorophenyl hydrazone (CCCP)
were added at indicated time points.
nature biotechnology VOLUME 28 NUMBER 3 MARCH 2010
Figure 5 Meclizine is cardioprotective in cellular and ex vivo models of
cardiac ischemia. (a) Protocol for the simulated ischemia-reperfusion
(SIR) injury model. (b) Viability of adult rat cardiomyocytes subjected to
SIR injury, in the presence of indicated concentrations of meclizine (Mec),
atropine (Atrop), pheniramine (Phenir), pyrilamine (Pyril) and scopolamine
(Scopo). (c) Respiration of cardiomyocytes after exposure to indicated
concentrations of meclizine. (d,e) Langendorff perfused rat hearts were
subjected to 25 min of global ischemia followed by 2 h of reperfusion.
Meclizine treatment comprised infusion of 1 µM meclizine from a port above the aortic cannula for 20 min, followed by a 1-min wash-out
before ischemia. (d) Rate pressure product (heart rate × left ventricular
developed pressure) is expressed as a percentage of the initial value
Cell viability (% of control)
throughout the ischemia-reperfusion (IR) protocol. (e) Following IR, hearts
were stained with TTC and infarct size was quantified. All data are
means ± s.e.m. from four to six individual experiments. (*, P < 0.05,
µM) µM) µM) µM) µM) µM) µM) µM)
ANOVA in d, Student's t-test in e).
Phenir (1 Scopo (1
a qualitative impact on membrane potential or redox potential during
respiratory state transitions of isolated mitochondria (.
Furthermore, meclizine treatment had no effect on mitochondrial morphology, membrane potential, mitochondrial (mt) DNA copy
number or the expression of mtRNAs in intact cells (Supplementary
(% of initial) 40
Fig. 6). Collectively, these observations demonstrate that unlike classic
Rate pressure product
inhibitors or uncouplers such as rotenone, antimycin, oligomycin
or carbonyl cyanide m-chlorophenyl hydrazone, meclizine does
IR alone IR + 1 µM
not itself directly inhibit and/or uncouple the OXPHOS machinery in isolated mitochondria, and does not reduce mitochondrial bio-
genesis in intact cells. Instead it may act through novel signaling or provide protection (). These results are consistent with the transcriptional mechanisms.
hypothesis that mild OXPHOS inhibition is cytoprotective in ischemic
Activation of HIF1-α or HIF2-α is known to induce transcriptional injury. As with the other cell types, meclizine inhibited oxygen
rewiring of energy metabolism from mitochondrial respiration to consumption in cardiomyocytes in a dose-dependent manner (
glycolysi. However, unlike with deferoxamine, a known inducer and Supplementary Fig. 8a,b) but not in isolated cardiac mitochondria
of the HIF pathway, we did not observe HIF1-α or HIF2-α stabiliza-
(Supplementary Fig. 8c,d). Next, we tested whether meclizine pro-
tion after meclizine treatment ( and Supplementary Fig. 7a,b). tects isolated, perfused rat hearts from ischemia-reperfusion injury in
Moreover, the kinetics of meclizine's OCR inhibition argue against a an ex vivo model of ischemic injury. Meclizine preserved heart pump transcriptional mechanism, as meclizine treatment resulted in inhibi-
function after the ischemic event () and significantly (P < 0.05)
tion within 2 h, whereas OCR inhibition mediated by deferoxamine reduced the infarct area of Langendorff perfused rat hearts subjected
only became apparent after 12 h (Supplementary Fig. 7c). In addition, to 25 min of global ischemia .
we examined the effect of meclizine treatment on HIF-responsive
Chemical preconditioning has also been shown to be protective in
genes using an HIF response element-luciferase reporter construct animal models of cerebral ischemia6,12,24–26. To determine whether
and recorded no induction of luciferase activity after a 6-h treatment meclizine might similarly be useful in this context, we first established
(Supplementary Fig. 7d).
safety and pharmacokinetic parameters for an intraperitoneal dosing
Collectively, these studies suggest that meclizine inhibits mitochon-
regimen (see Online Methods). We found that mice tolerate daily
drial respiration indirectly, in an HIF-independent manner that does intraperitoneal injections of 100 mg/kg meclizine without any weight not involve histaminergic or muscarinic receptor signaling.
loss or behavioral changes even after four consecutive days. Six hours after a single intraperitoneal dose, the plasma concentration is in the
Meclizine pretreatment confers protection against ischemic injury
3–5 µM range, a concentration sufficient to attenuate mitochondrial
Previous studies have clearly established that brief, nonlethal respiration of primary mouse neurons (Supplementary Fig. 9). We
episodes of ischemia can confer prophylaxis against subsequent then tested whether meclizine protects against cerebral ischemia by
stroke or myocardial infarction, and studies in model systems have pretreating mice with two intraperitoneal injections of 100 mg/kg
shown that chemical inhibition of mitochondrial respiration can meclizine, or an equal volume of vehicle at 17 h and 3 h before a 1 h
mimic this protection, a process coined "chemical preconditioning. transient middle cerebral artery occlusion (). We found that
Having shown that meclizine, an over-the-counter drug that crosses total infarct volume was significantly reduced by 23% in meclizine-
the blood-brain barrier can attenuate mitochondrial respiration, we treated animals (P = 0.03, . In addition, meclizine signifi-
sought to determine whether this drug is cardioprotective and neuro-
cantly reduced the area of infarction in brain slices with the greatest
protective in cellular and animal models.
area of infarct (P = 0.02, . The in vivo protective effect
First, we tested meclizine in an adult rat ventricular cardiomyocyte of meclizine is likely independent of its antihistamine or antimus-
model of simulated ischemia-reperfusion injury (). A 20-min carinic property, as treatment with pyrilamine or scopolamine did meclizine pre-incubation followed by wash-out before ischemia not decrease infarct volume ( or reduce the area of infarction elicited a dose-dependent protection of cardiomyocytes against cell in brain slices . Furthermore, meclizine-pretreated animals death, whereas other antihistamines (pyrilamine and pheniramine) tended toward having preserved neurological function compared and antimuscarinic agents (scopolamine and atropine) did not to controls (P = 0.07, Kruskal-Wallis non-parametric ANOVA).
VOLUME 28 NUMBER 3 MARCH 2010 nature biotechnology
Figure 6 Meclizine is neuroprotective in a mouse model of stroke.
(a) Protocol for the murine model of stroke. Male C57BL/6 mice were
treated with two intraperitoneal injections of 100 mg/kg meclizine,
20 mg/kg pyrilamine and 0.5 mg/kg scopolamine or vehicle at 17 h and
injection injection
3 h before 1 h transient middle cerebral artery occlusion followed by 24 h
of reperfusion. (b) Cerebral blood flow (CBF) measured at baseline and
after occlusion of the common carotid artery (CCA) and middle cerebral
artery (MCA) upon treatment with meclizine, scopolamine, pyrilamine
CBF (% of baseline)
or vehicle. Data represent mean ± s.d. (c) Infarct volume measured
–10 0 10 20 30 40 50 60 70 80
on TTC-stained 1-mm thick coronal slices obtained from mice treated
with meclizine, scopolamine, pyrilamine or vehicle. Data points refer to
independent experiments, and the solid line represents their mean.
(*P < 0.05 versus vehicle and scopolamine, P < 0.01 versus pyrilamine;
one-way ANOVA followed by Tukey's multiple comparison test).
(d) Representative images of TTC-stained 1-mm thick coronal brain
sections (slice 1–10). (e) Infarct area in the rostrocaudal extent of
the brain (slice 1–10) upon treatment with meclizine, scopolamine,
pyrilamine or vehicle. Data points represent the mean area of infarction
in individual slice levels ± s.d. in mm2 (n = 14 for vehicle, n = 8 for
meclizine, n = 8 for pyrilamine, n = 5 for scopolamine, *P < 0.05).
Slice numbers 1 – 10
hydroxylase 1 knockout mice are resistant to acute ischemia because
of reduced generation of oxidative stress In preclinical studies, pro-lyl hydroxylase inhibitors confer protection in models of myocardial
infarctio, strok and renal ischemi. However, as HIF regulates the expression of a plethora of genes, and unwanted side effects have
remained a concer, it might be useful to expand the arsenal of agents that shift energy metabolism.
Our screen has identified a new metabolic activity for meclizine,
an over-the-counter drug that has been in use in the United States for
The cerebral blood flow deficit and the amount of post-
> 40 years for treatment of nausea and vertigo. We found that 1 µM
operative weight loss did not differ between the groups.
meclizine pretreatment provided cytoprotection in in vitro and ex vivo models of cardiac ischemia-reperfusion injury ( In addition, we
showed that prophylaxis with meclizine significantly reduced infarct
Recent studies have shown that changes in cellular energy metabolism volume in an in vivo model of cerebral ischemia (. The utility
can accompany a range of human diseases, and that targeting energy of pretreatment paradigms described in this study arises in clinical metabolism may hold therapeutic potential. However, we lack an arse-
settings in which ischemic insults can be anticipated. Examples of
nal of clinically safe and useful agents that target energy metabolism such situations include patients undergoing high-risk surgical pro-by distinct mechanisms. In this study, we have introduced a facile, cedures and the large cohort of patients that suffer from diseases of
nutrient-sensitized screening strategy aimed at identifying small recurrent ischemia, such as unstable angina or recurrent transient molecules that shift cellular energy metabolism from mitochondrial ischemic attacks An open question is whether currently approved respiration to glycolysis. We have identified several FDA-approved doses of meclizine achieve the required blood concentrations required drugs that exhibit such activity and may have potential for helping to for cardioprotection or neuroprotection. Post-marketing surveillance abrogate ischemia-reperfusion injury in the heart, brain and perhaps data support the safe nonprescription use of meclizine, and published other sensitive organ systems such as the lung or kidney. Focusing on studies in animals, including nonhuman primates, have shown that one specific hit from our screen, meclizine, we have demonstrated that higher doses can be tolerated39,40. However, because the potency of it suppresses OXPHOS by means of a mechanism distinct from classic meclizine in attenuating mitochondrial respiration appears to vary inhibitors or uncouplers, and that it confers protection against cardiac
across cell types (Figs. 3c and 5c and Supplementary Fig. 9), pre-
and cerebral ischemia in cellular and animal models.
clinical studies of efficacy and toxicity are required to rigorously
A large body of literature demonstrates that agents that blunt determine optimal dosing and safety regimens before evaluating the
mitochondrial respiration can offer prophylaxis against cell death utility of meclizine for new clinical indications in humans.
after ischemia and reperfusion in the heart4,5,27 or brain3,6,26. This
Our detailed studies on the effects of meclizine on cellular energy
effect is thought to occur through suppressing oxidative injury, and metabolism clearly show that it attenuates mitochondrial respiration in
may be related to protection conferred by ischemic preconditioning, a manner distinct from other drugs of known mechanism of action and
although the precise molecular mechanism is not known. Notably, independent of the HIF pathway ( and Supplementary Fig. 7).
redirecting energy metabolism toward glycolysis can reduce oxida-
In contrast to canonical inhibitors, meclizine does not directly target
tive damage and suppresses apoptosis28–30. Interestingly, switching to the OXPHOS machinery in isolated mitochondria ( and can be
anaerobic metabolism appears to be a natural adaptation to reduced titrated over a broad range of concentrations to achieve inhibition of
oxygen availability and activation of the HIF pathway provides one cellular OCR by 10–60% (). Our data suggest that meclizine
such strategy for redirecting energy metabolism toward glycolysis9,32. acts independently of the muscarinic or histamine receptors, as drugs
Recent studies using genetic and chemical approaches of activating affecting these two receptors did not inhibit OCR (Supplementary Fig. 5)
the HIF pathway have shown promising results in various models and they do not confer neuroprotection or cytoprotection in our
of ischemia-reperfusion injury. For example, myofibers of prolyl models (Figs. 5 and 6). We do not know the precise molecular target
nature biotechnology VOLUME 28 NUMBER 3 MARCH 2010
of meclizine responsible for this effect on energy metabolism, toxicity data; S. Calvo, A. Chess, R. Gould, E. Lander, A. Ting, S. Vafai and members but one possibility is a metabolic target outside the mitochon-
of Mootha lab for valuable discussions and comments. This work was supported
drion whose subsequent impact is to direct metabolism away from by fellowships or grants from the United Mitochondrial Disease Foundation
(V.M.G.); Howard Hughes Medical Institute (S.A.S. and V.K.M.); National
OXPHOS. Alternatively, meclizine may undergo a bio-transformation Institutes of Health (RO1 HL-071158 to P.S.B.); Deane Institute for Integrative
into a product that directly inhibits the OXPHOS machinery. We Research in Stroke and Atrial Fibrillation (C.A.); American Heart Association cannot exclude the possibility that meclizine, like many clinically used (#0815770D to A.P.W.); the Burroughs Wellcome Fund (V.K.M.); the Center for drugs, hits multiple cellular targets to affect cellular energetics and Integration of Medical and Innovative Technology (V.K.M.); and the American
Diabetes Association/Smith Family Foundation (V.K.M.).
Nutrient-sensitized screening, as we have presented it, builds on AUtHoR CoNtRIBUtIoNS
previous studies that have shown that many cultured cells generate V.M.G. and V.K.M. conceived the project; V.M.G., S.A.S., J.H.L., W.C., F.P., C.B.C. their ATP from either glycolysis or glutamine oxidation14,17. However, and A.P.W. performed experiments; V.M.G., S.A.S., J.H.L., R.N., F.P., C.A., P.S.B. the screening strategy may not necessarily work in other cell types, and V.K.M. performed statistical and data analysis; V.M.G., S.A.S. and V.K.M.
wrote the paper.
for example, cells with limited metabolic flexibility, cells that do not
have pathways for glutamine oxidation, or postmitotic cells in which CoMPetING INteReStS StAteMeNt
a growth assay is not possible. Another limitation of our approach The authors declare competing financial interests: details accompany the full-text
is that the compounds that emerge from the screen may act not only HTML version of the paper at
on energy-related pathways, but also potentially on other properties Published online at http://www.nature.com/naturebiotechnology/.
influenced by the switch in nutrients. For example, we have noted reprints and permissions information is available online at http://npg.nature.com/that cells grown in glucose tend to proliferate more quickly, and for reprintsandpermissions/.
this reason, drugs from the right side of the tail ( could either
be blunting glycolytic metabolism or affecting rapid proliferation. 1. Warburg, O. On the origin of cancer cells. Science 123, 309–314 (1956).
Hence, secondary assays are still required to confirm the energetic 2. Bonnet, S. et al. A mitochondria-K+ channel axis is suppressed in cancer and its
normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 11,
consequences of a drug identified by our screening assay.
37–51 (2007).
Our screen contained only a few thousand compounds and has 3. Huber, R., Spiegel, T., Buchner, M. & Riepe, M.W. Graded reoxygenation with
already shown high sensitivity for identifying drugs that target
chemical inhibition of oxidative phosphorylation improves posthypoxic recovery in
murine hippocampal slices. J. Neurosci. Res. 75, 441–449 (2004).
cellular pathways of energy metabolism. As we have shown here, com-
4. Burwell, L.S., Nadtochiy, S.M. & Brookes, P.S. Cardioprotection by metabolic shut-
pounds emerging from the upper tail of the distribution (
down and gradual wake-up. J. Mol. Cell. Cardiol. 46, 804–810 (2009).
5. Chen, Q., Camara, A.K., Stowe, D.F., Hoppel, C.L. & Lesnefsky, E.J. Modulation of
and Supplementary Tables 2 and 4) could serve as valuable lead com-
electron transport protects cardiac mitochondria and decreases myocardial injury
pounds for prophylaxis against heart attack and stroke, but they may
during ischemia and reperfusion. Am. J. Physiol. Cell Physiol. 292, C137–C147
also find broader application for preventing or treating a wide variety
6. Riepe, M.W. et al. Increased hypoxic tolerance by chemical inhibition of oxidative
of other diseases involving oxidative damage, such as neurodegenera-
phosphorylation: "chemical preconditioning". J. Cereb. Blood Flow Metab. 17,
tive disorders. The opposite tail includes dozens of compounds
257–264 (1997).
already used as chemotherapeutic agents, perhaps due to their selec-
7. Piantadosi, C.A. & Zhang, J. Mitochondrial generation of reactive oxygen species
after brain ischemia in the rat. Stroke 27, 327–331 (1996).
tive toxicity in more rapidly proliferating cells, and may include addi-
8. Kaelin, W.G. Jr. & Ratcliffe, P.J. Oxygen sensing by metazoans: the central role of
tional, clinically safe agents that could be useful as adjuvant therapies.
the HIF hydroxylase pathway. Mol. Cell 30, 393–402 (2008).
9. Kim, J.W., Tchernyshyov, I., Semenza, G.L. & Dang, C.V. HIF-1-mediated expression
The results of our screen may help to pinpoint clinical benefits (or
of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation
toxicity) of drugs that are not readily attributable to their known
to hypoxia. Cell Metab. 3, 177–185 (2006).
targets or mechanism of action. We anticipate that this strategy can 10. Fukuda, R. et al. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency
of respiration in hypoxic cells. Cell 129, 111–122 (2007).
be extended to other nutrients—such as fatty acids or ketone bodies. 11. Semenza, G.L. Defining the role of hypoxia-inducible factor 1 in cancer biology The nutrient-sensitized assay can also be used to screen a much larger
and therapeutics. Oncogene published online, doi:10.1038/onc.2009.441
library of compounds or even genome-wide RNA interference pertur-
(30 November 2009).
12. Fraisl, P., Aragones, J. & Carmeliet, P. Inhibition of oxygen sensors as a therapeutic
bations to systematically understand drug action and gene function
strategy for ischaemic and inflammatory disease. Nat. Rev. Drug Discov. 8, 139–152
within the broader context of cellular energy homeostasis.
13. Hoesch, R.E. & Geocadin, R.G. Therapeutic hypothermia for global and focal
ischemic brain injury–a cool way to improve neurologic outcomes. Neurologist 13,
331–342 (2007).
Methods and any associated references are available in the online 14. Reitzer, L.J., Wice, B.M. & Kennell, D. Evidence that glutamine, not sugar, is the
major energy source for cultured HeLa cells. J. Biol. Chem. 254, 2669–2676
version of the paper at http://www.nature.com/naturebiotechnology/.
15. Robinson, B.H., Petrova-Benedict, R., Buncic, J.R. & Wallace, D.C. Nonviability of
Availability of raw data. Screening data in the form of cell count per
cells with oxidative defects in galactose medium: a screening test for affected
patient fibroblasts. Biochem. Med. Metab. Biol. 48, 122–126 (1992).
well are available at ChemBank for galactose plates 16. Marroquin, L.D., Hynes, J., Dykens, J.A., Jamieson, J.D. & Will, Y. Circumventing and glucose
the Crabtree effect: replacing media glucose with galactose increases susceptibility
of HepG2 cells to mitochondrial toxicants. Toxicol. Sci. 97, 539–547 (2007).
17. DeBerardinis, R.J. et al. Beyond aerobic glycolysis: transformed cells can engage
in glutamine metabolism that exceeds the requirement for protein and nucleotide
synthesis. Proc. Natl. Acad. Sci. USA 104, 19345–19350 (2007).
Note: Supplementary information is available on the website.
18. Wu, M. et al. Multiparameter metabolic analysis reveals a close link between
attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency
in human tumor cells. Am. J. Physiol. Cell Physiol. 292, C125–C136 (2007).
We thank E. Shoubridge for the MCH58 cell line; M. MacDonald for immortalized
19. Wagner, B.K. et al. Large-scale chemical dissection of mitochondrial function. Nat.
Biotechnol. 26, 343–351 (2008).
striatal cells; R. Xavier for the HRE luciferase construct; S. Norton, B. Wagner and
20. Golenser, J., Waknine, J.H., Krugliak, M., Hunt, N.H. & Grau, G.E. Current
the Broad Chemical Screening Platform for assistance in compound arraying;
perspectives on the mechanism of action of artemisinins. Int. J. Parasitol. 36,
J. Evans of the Whitehead Institute/MIT BioImaging Center for assistance with
1427–1441 (2006).
high-throughput microscopy; C. Belcher-Timme for technical assistance; T. Kitami
21. The Food and Drug Administration Antiemetic drug products for over-the-counter
for assistance with mitochondrial imaging; M. Mehta for assistance reviewing drug
human use; final monograph. Fed. Regist. 52, 15866–15893 (1987).
VOLUME 28 NUMBER 3 MARCH 2010 nature biotechnology
22. Brunton, L.L., Lazo, J.S. & Parker, K.L. Goodman & Gilman's The Pharmacological
32. Lu, C.W., Lin, S.C., Chen, K.F., Lai, Y.Y. & Tsai, S.J. Induction of pyruvate
Basis of Therapeutics, edn. 11 (The McGraw-Hill Companies, 2006).
dehydrogenase kinase-3 by hypoxia-inducible factor-1 promotes metabolic switch
23. Papandreou, I., Cairns, R.A., Fontana, L., Lim, A.L. & Denko, N.C. HIF-1 mediates
and drug resistance. J. Biol. Chem. 283, 28106–28114 (2008).
adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption.
33. Aragones, J. et al. Deficiency or inhibition of oxygen sensor Phd1 induces hypoxia
Cell Metab. 3, 187–197 (2006).
tolerance by reprogramming basal metabolism. Nat. Genet. 40, 170–180
24. Gidday, J.M. Cerebral preconditioning and ischaemic tolerance. Nat. Rev. Neurosci.
7, 437–448 (2006).
34. Philipp, S. et al. Stabilization of hypoxia inducible factor rather than modulation
25. Sugino, T., Nozaki, K., Takagi, Y. & Hashimoto, N. 3-Nitropropionic acid induces
of collagen metabolism improves cardiac function after acute myocardial infarction
ischemic tolerance in gerbil hippocampus in vivo. Neurosci. Lett. 259, 9–12 (1999).
in rats. Eur. J. Heart Fail. 8, 347–354 (2006).
26. Ratan, R.R. et al. Translation of ischemic preconditioning to the patient: prolyl
35. Siddiq, A. et al. Hypoxia-inducible factor prolyl 4-hydroxylase inhibition. A target
hydroxylase inhibition and hypoxia inducible factor-1 as novel targets for stroke
for neuroprotection in the central nervous system. J. Biol. Chem. 280,
therapy. Stroke 35, 2687–2689 (2004).
27. Lesnefsky, E.J. et al. Blockade of electron transport during ischemia protects cardiac
36. Bernhardt, W.M. et al. Preconditional activation of hypoxia-inducible factors
mitochondria. J. Biol. Chem. 279, 47961–47967 (2004).
ameliorates ischemic acute renal failure. J. Am. Soc. Nephrol. 17, 1970–1978
28. Jeong, D.W., Kim, T.S., Cho, I.T. & Kim, I.Y. Modification of glycolysis affects cell
sensitivity to apoptosis induced by oxidative stress and mediated by mitochondria.
37. Brahimi-Horn, M.C. & Pouyssegur, J. Harnessing the hypoxia-inducible factor in
Biochem. Biophys. Res. Commun. 313, 984–991 (2004).
cancer and ischemic disease. Biochem. Pharmacol. 73, 450–457
29. Hunter, A.J., Hendrikse, A.S. & Renan, M.J. Can radiation-induced apoptosis be
modulated by inhibitors of energy metabolism? Int. J. Radiat. Biol. 83, 105–114
38. Dirnagl, U., Becker, K. & Meisel, A. Preconditioning and tolerance against cerebral
ischaemia: from experimental strategies to clinical use. Lancet Neurol. 8, 398–412
30. Vaughn, A.E. & Deshmukh, M. Glucose metabolism inhibits apoptosis in neurons
and cancer cells by redox inactivation of cytochrome c. Nat. Cell Biol. 10,
39. Giurgea, M. & Puigdevall, J. Experimental teratology with Meclozine. Med.
1477–1483 (2008).
Pharmacol. 15, 375–388 (1966).
31. Ramirez, J.M., Folkow, L.P. & Blix, A.S. Hypoxia tolerance in mammals and birds:
40. Lione, A. & Scialli, A.R. The developmental toxicity of the H1 histamine antagonists.
from the wilderness to the clinic. Annu. Rev. Physiol. 69, 113–143 (2007).
Reprod. Toxicol. 10, 247–255 (1996).
nature biotechnology VOLUME 28 NUMBER 3 MARCH 2010
package CellProfiler. Images were analyzed individually by first identifying
All experiments were done in accordance with the national and institutional
nuclei using the Hoechst staining signal. Total number of nuclei per field were
guidelines for animal welfare, adhering to protocols approved by the institu-
recorded, and the sum of nuclei from four images per well gave cell count
tional subcommittee on research animal care.
per well. Computation was performed using a UNIX computer cluster. The unprocessed data from our screen in the form of cell count per well are avail-
Cell culture. Immortalized MCH58 human diploid fibroblasts containing the
able in ChemBank. Please note that the ChemBank data sets include statistical
pLKO.1 vector were grown in DMEM high-glucose medium (Invitrogen) with
calculations that were not used in our final analysis. For a detailed description
10% FBS (Sigma), 1× penicillin, streptomycin and glutamine (Invitrogen),
of our statistical methods, please refer to the section below "Statistical analysis
2 µl/ml puromycin and 50 µg/ml uridine at 37 °C and 5% CO2. The high- of screening data," which outlines the method used to calculate our summary glucose medium was replaced with 10 mM glucose or 10 mM galactose wherever
statistic, Sglu/gal. The Sglu/gal value for every compound included in our screen
indicated. All media contained 1 mM pyruvate and 4 mM glutamine. MCH58
can be found in Supplementary Table 1.
fibroblasts (without pLKO.1), HeLa and HEK293 cells were grown in DMEM
high-glucose medium with 10% FBS at 37 °C and 5% CO2; STHdhQ7/7 mouse Assays of mitochondrial physiology. Mitochondria were isolated from
striatal cells were grown at 33 °C.
C57BL/6 mouse kidneys by differential centrifugation and resuspended in experimental buffer as described previously, to a final concentration of
Measurement of cellular OCR and ECAR. OCR and ECAR measurements
0.5 mg/ml. State 2 respiration was initiated with the addition of 12.5 mM
were carried out as previously describe with minor modifications. Briefly,
glutamate and 12.5 mM malate, or 20 mM pyruvate and 1 mM malate or
MCH58 fibroblasts were seeded in XF24-well cell culture microplates (Seahorse
12 mM succinate and 3 µM rotenone. State 3 respiration was initiated with
Bioscience) at 30,000 cells/well in 10 mM glucose- or 10 mM galactose–
the addition of 0.2 mM ADP, and uncoupled respiration was initiated with the
containing media and incubated at 37 °C and 5% CO2 for 20 h. Before the addition of 5 µM carbonyl cyanide m-chlorophenyl hydrazone. Mitochondria measurements were made, the growth medium was replaced with 925 µl of
were incubated for 5 min in respiration buffer pH 7.4 supplemented with
assay medium (with 10 mM glucose or 10 mM galactose as the sugar source)
either meclizine (50 µM) or DMSO. O2 consumption was monitored with a
and cells were incubated at 37 °C for 60 min. The OCR and ECAR measure-
Fiber Optic Oxygen sensor probe (Ocean Optics) at 25 °C, and NADH (endog-
ments on HeLa, HEK293 and STHdhQ7/7 mouse striatal cells were carried out
enous, 370 ± 7 nm excitation, 440 ± 4 nm emission) and membrane potential
by growing them in XF24 plates at 40,000/well (HeLa and STHdhQ7/7 mouse
(1.25 µM TMRM, 546 ± 7 nm excitation, 590 ± 4 nm emission) were measured
striatal cells) or 50,000/well (HEK293 cells) for 20 h under their regular
with a Perkin-Elmer LS50B luminescence spectrometer.
growth conditions, as described in the previous section. The assay medium was
the same as above except 25 mM glucose was used as the sugar source. Three
Drug testing in cardiac ischemia-reperfusion injury. Adult rat ventricular
baseline measurements were recorded before the addition of compounds. For
cardiomyocytes were isolated by endotoxin-free collagenase perfusion, and
the secondary screening assay on selected compounds listed in ,
the simulated ischemia-reperfusion injury was performed as previously
MCH58 fibroblasts were grown in 25 mM glucose-containing medium in the
describe. Briefly, ischemia comprised 1 h in anoxic glucose-free buffer at
presence of a compound for 16–20 h. Each compound was tested at screening
pH 6.5, and reperfusion comprised 30 min in normoxic glucose-replete buffer
concentration—antimycin (3.65 µM), ascorbate (10 µM), clofilium tosylate
at pH 7.4. Cell viability was monitored by Trypan blue exclusion. Rat cardiac
(5.9 µM), nifuroxazide (7.27 µM), meclizine (10 µM), menadione (11.62 µM),
mitochondria were isolated, and respiration of both cells and mitochondria
clemastine (5.82 µM), vinpocetine (5.71 µM), bisacodyl (5.53 µM), nonoxynol-9
was measured using a Clark oxygen electrode, as previously described
(10 µM), sertraline (10 µM), thonzonium (3.91 µM), chlorhexidine (3.96 µM),
Isolated rat hearts were retrograde reperfused in Langendorff mode under
mefloquine (10 µM) and alexidine (3.93 µM). OCR measurements on the
constant flow as described previousl.
mouse primary cortical neurons obtained from day E14–15 embryos were carried out in their regular growth medium.
Drug testing in neuronal ischemia-reperfusion injury. Plasma concentra-
tions of meclizine were determined after intraperitoneal injections in C57BL/6
Cell viability assays. MCH58 fibroblasts were seeded in 96-well plates at 5,000
mice. Injections of 100 mg/kg meclizine or vehicle control were followed
cells/well in DMEM high-glucose medium. After 20 h, cells were washed in
by cardiac puncture blood draws at 1 h or 6 h. Absolute concentrations of
PBS and the growth medium was replaced with 10 mM glucose- or 10 mM
meclizine in plasma were measured using liquid chromatography tandem mass
galactose-containing media containing different concentrations of meclizine,
spectrometry against a purified standard. To check the protective effect of the
rotenone, antimycin, oligomycin or DMSO (0.1%). The cells were then grown
tested compounds, male C57BL/6 mice were treated with two intraperitoneal
for 72 h and cell viability was assayed by the CellTiter-Glo Luminescent
injections of 100 mg/kg meclizine, 20 mg/kg pyrilamine and 0.5 mg/kg sco-
Viability assay (Promega).
polamine or vehicle, 17 h and 3 h before ischemia. Drug doses for pyrilamine and scopolamine were chosen based on previous literature evidence of in vivo
Chemical screening and high-throughput assay of cell number quantifica-
brain bioavailability46,47. The experimenter was blinded to treatment groups.
tion. MCH58 fibroblasts were seeded at 5,000 cells/well using the robotic
Mice were anesthetized with isoflurane (2.5% induction, 1.5% maintenance,
MultiDrop Combi (ThermoFisher Scientific) dispenser into 96-well plates
in 70% N2O/30% O2), and subjected to 1 h transient middle cerebral artery
(PerkinElmer) at 100 µl per well in DMEM high-glucose medium. Twenty-four
occlusion using an intraluminal filament inserted through the external carotid
hours later, cells were washed twice in PBS and medium was replaced with
artery. Regional cerebral blood flow was monitored using a laser Doppler probe
either 10 mM glucose- or 10 mM galactose-containing media. Approximately
placed over the core middle cerebral artery territory. Rectal temperature was
100 nl of each compound was pin-transferred in duplicate into the plates with
controlled at 37 °C by a servo-controlled heating pad. Total infarct volume
a steel pin array using the CyBi-Well robot (CyBio). The compound collec-
was calculated on 2,3,5-triphenyltetrazolium chloride (TTC)-stained 1-mm
tion of 3,695 drugs includes two commercially available libraries (Spectrum
thick coronal sections by integrating the infarct areas in each of ten slice levels.
and Prestwick). Compound-treated plates were incubated at 37 °C for 72 h.
Infarct volume was calculated using the ‘indirect method', that is, contralateral
Cells were then washed once with PBS, stained with 10 µM Hoechst 33342
hemisphere minus ipsilateral non-infarcted volume. Data were expressed as
(Invitrogen) and fixed in 3.7% formaldehyde solution for 15 min. Wells were
mean ± s.d. One-way ANOVA followed by Tukey's multiple comparison test
then washed once and stored in PBS. Cell culture plates were stored at 4 °C
was used for analysis of values between groups. Neurological deficit scores
until the time of imaging, which was at most 24 h after fixation. Imaging
were analyzed by Kruskal-Wallis nonparametric ANOVA followed by Dunn's
was performed by Arrayscan VTi automated microscope (ThermoFisher
multiple comparisons test. P < 0.05 was considered statistically significant.
Scientific) with the use of an automated plate stacker at the Whitehead Institute Biomedical Imaging Center. Four nonoverlapping images at 5× magnification
Western blot analysis. HIF1-α and HIF2-α stabilization was assessed in
(NA 0.25) were acquired for a field of view of 1.3 mm × 1.3 mm per image.
MCH58 fibroblasts and HeLa cells by carrying out western blot detection using
Image analysis was performed using the freely available open-source software
anti-HIF1-α (Cell Signaling) and anti-HIF2-α (Novus Biologicals) antibodies.
Protein was extracted from cells pretreated with either 0.1% DMSO, 50 µM
well with the trimmed mean (the average after discarding the largest and
meclizine or 100 µM deferoxamine (Sigma). SDS PAGE was performed on
smallest value) of the cell counts for the 16 DMSO-treated wells on each
15 µg/lane protein using NuPAGE 4–12% Bis-Tris gel from Invitrogen. Western
plate. A single 96-well plate exhibited very low counts in all wells and was
blot analysis was performed as per the standard procedures. β-actin was used
discarded. We then removed measurements that failed to replicate by requir-
as a loading control.
ing that the ratio of normalized counts was <1.5 between replicates; this excluded 49 wells. The remaining replicate measurements were averaged,
HIF reporter assay. Luciferase activity in HeLa cells transiently transfected
and fold changes were computed as the ratio between the averaged normal-
with HIF response element (HRE)/luciferase construct was measured by the
ized counts for the glucose and galactose screens. The Sglu/gal for each com-
Dual-Luciferase Reporter Assay System (Promega). The drug treatment was
pound was calculated by taking the log10 of the normalized and averaged fold
initiated 24 h after the transfection and the treatment was continued for
change in glucose divided by galactose. To evaluate statistical significance,
6 h with 0.1% DMSO, 100 µM deferoxamine or 50 µM meclizine. HeLa cells
we computed Z-scores for each well against the DMSO (null) distribution
were transfected using the Fugene reagent (Roche) as per the manufacturer's
and averaged Z-scores across replicates. Wells with average Z-score > 2.5
instruction. HRE reporter construct was a generous gift from R. Xavier,
were considered significant.
Massachusetts General Hospital.
Assays of mitochondrial abundance, morphology and membrane potential
41. Carpenter, A.E. et al. CellProfiler: image analysis software for identifying and
in intact cells. Mitochondrial (mt) morphology, membrane potential, mtDNA
quantifying cell phenotypes. Genome Biol. 7, R100 (2006).
content and mtDNA expression were measured in HeLa cells treated with 50 µM
42. Mootha, V.K., Arai, A.E. & Balaban, R.S. Maximum oxidative phosphorylation
of meclizine over a 6-h period. MitoTracker CMXRos (Invitrogen) and Hoechst
capacity of the mammalian heart. Am. J. Physiol. 272, H769–H775 (1997).
43. Wojtovich, A.P. & Brookes, P.S. The complex II inhibitor atpenin A5 protects against
33342 staining of live cells was performed as per manufacturer's recommenda-
cardiac ischemia-reperfusion injury via activation of mitochondrial KATP channels.
tion. The stained cells were observed at 20× magnification with an Olympus
Basic Res. Cardiol. 104, 121–129 (2009).
CKX41 microscope and fluorescent images were captured with a QiCAM camera.
44. Wojtovich, A.P. & Brookes, P.S. The endogenous mitochondrial complex II inhibitor
The mitochondrial membrane potential was measured with a ratiometric dye,
malonate regulates mitochondrial ATP-sensitive potassium channels: implications
for ischemic preconditioning. Biochim. Biophys. Acta 1777, 882–889 (2008).
JC-1 by measuring relative fluorescent units at 590 nm and 535 nm using a
45. Nadtochiy, S.M., Tompkins, A.J. & Brookes, P.S. Different mechanisms of
fluorescent plate reader. The mtDNA copy number and mtRNA transcripts were
mitochondrial proton leak in ischaemia/reperfusion injury and preconditioning:
measured by quantitative real-time PCR assays as described previously.
implications for pathology and cardioprotection. Biochem. J. 395, 611–618
(2006).
46. Miyazaki, S., Imaizumi, M. & Onodera, K. Effects of thioperamide, a histamine
Drug annotation. All FDA-approved drugs were annotated after confirming
H3-receptor antagonist, on a scopolamine-induced learning deficit using an elevated
their entry in the Orange book, 29th edition 2009. For other clinically used
plus-maze test in mice. Life Sci. 57, 2137–2144 (1995).
compounds, we used the corresponding PubMed and PubChem entries.
47. Toyota, H. et al. Behavioral characterization of mice lacking histamine H(3)
receptors. Mol. Pharmacol. 62, 389–397 (2002).
48. Baughman, J.M. et al. A computational screen for regulators of oxidative
Statistical analysis of screening data. Normalized cell counts were com-
phosphorylation implicates SLIRP in mitochondrial RNA homeostasis. PLoS Genet.
puted separately for each 96-well plate by dividing the cell count of each
5, e1000590 (2009).
Source: http://www.perocchi.genzentrum.lmu.de/assets/Uploads/Gohil-2010.pdf
HKFRS 6 and HKAS 41 Nelson Lam CFA FCCA FCPA(Practising)MBA MSc BBA CPA(US) ACA xploration fo r & Evaluation of r & Evaluation of Mineral Reso urce Exploration for and Evaluation of Mineral Resources (HKFRS 6) Exploration for and Evaluation of Mineral Resources – Sharing Points Objective of HKFRS 6 Limited Improvements
ERSTE The ESG Letter January, February/2013 Edition RESPONSIBLE RETURN INVESTMENT BOARDHealthcare provision – a basic need not always met ENGAGEMENTAccess to Medicine COMPANY OF THE MONTHGlaxoSmithKline PLC PIN IT DOWNBitter pills & healthy capitalists? ERSTE ASSET MANAGEMENT 02 When we wish each other a Happy New Year – as was the case just a few days ago – then the topic of health will almost always be at the forefront of our minds. As human beings, being healthy and staying healthy is accorded a high priority. The necessary healthcare provision is good to very good in large parts of Europe, despite the fact that even in these countries a debate about a „two-tier society in medical care" keeps emerging from time to time.